International Journal of Epidemiology And Public Health Research
OPEN ACCESS | Volume 9 - Issue 1 - 2026
ISSN No: 2836-2810 | Journal DOI: 10.61148/2836-2810/IJEPHR


Tewodros Legesse
Department of microbiology immunology and veterinary public health MSc on veterinary public health; Addis Ababa University College of Veterinary Medicine and Agriculture.
*Corresponding author: Tewodros Legesse, Department of microbiology immunology and veterinary public health MSc on veterinary public health; Addis Ababa University College of Veterinary Medicine and Agriculture.
Received: April 25, 2025
Accepted: May 03, 2025
Published: May 07, 2025
Citation: Tewodros Legesse. (2025) “Review On Evidence Of Zoonotic Origin Of Human Coronaviruses”. International Journal of Epidemiology and Public Health Research, 6(3); DOI: 10.61148/2836- 2810/IJEPHR/125
Copyright: © 2025. Tewodros Legesse. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited., provided the original work is properly cited.
The Coronaviruses (CoVs) are the largest group of viruses belonging to the Nidovirales order, which includes Coronaviridae, Arteriviridae, Mesoniviridae, and Roniviridae families. They all contain very large genomes for RNA viruses, with some viruses having the largest identified RNA genomes, containing up to 33.5 kilobase (kb) genomes. Coronaviruses are enveloped, non-segmented, positive-sense RNA viruses that are known to infect humans and a wide variety of animals, causing mainly respiratory diseases in humans, although other organ systems are also affected with varying severity. Two highly pathogenic coronaviruses have claimed thousands of lives globally in the past two decades, leading to renewed interest in the study of their evolution, transmission and pathogenicity. Severe acute respiratory syndrome coronavirus (SARS-CoV) emerged in southern China in 2002/2003 and spread rapidly to cause a global pandemic, leading to more than 8000 confirmed cases of SARS with a case fatality rate of nearly 10%. Almost 10 years after the SARS outbreak, Middle East respiratory syndrome coronavirus (MERS-CoV) emerged as a highly fatal human pathogen in the Arabian Peninsula in 2012 with even higher mortality approaching 40%. Before the SARS-CoV pandemic, only two human coronaviruses (HCoVs) were known, namely HCoV-229E and HCoV-OC43. HCoVs typically cause mild, self-limiting upper respiratory infections in humans, although occasional cases of severe lower respiratory infection have been reported. Over the past two decades, studies in searching for the zoonotic origin of SARS-CoV and MERS-CoV have seen breakthrough. Several zoonotic spillover infections or epidemics in humans are explained by the involvement of intermediate hosts, creating additional complexity when analyzing multi host species of viruses. Intermediate hosts may act as zoonotic sources for dead-end hosts not only because they close gaps of contact between species. Therapies and preventive strategies such as vaccination are generally limited for these emerging zoonotic pathogens and no treatment options for fatal human infections. This paper tries to review briefly on the zoonotic origin of human coronavirus.
1. Introduction:
Coronaviruses are a large family of viruses that cause a range of illnesses in humans, from the common cold to the severe respiratory syndrome (SARS) viruses in this family also cause a few animal diseases (WHO, 2014). The name “coronavirus” is derived from the Latin corona, meaning crown or halo, and refers to the characteristic’s appearance of virions. This morphology is created by the viral S peplomers, which are proteins that populate the surface of the virus and determine host tropism. Coronavirus particles are irregularly shaped, approximately 60-220 nm in diameter, with an outer envelope bearing distinctive, ‘club-shaped’ peplomers (de Groot et al., 2011).
Several zoonotic spillover infections or epidemics in humans are explained by the involvement of intermediate hosts, creating additional complexity when analyzing multi host species of viruses. The viruses existing in the primordial host and the intermediate host can both belong to the same species, but the viral source population involved in zoonotic transmission may be disconnected from that in the primordial host. Intermediate hosts may act as zoonotic sources for dead-end hosts not only because they close gaps of contact between species, but also because they could make the virus transmissible by intermediary adaptation (Caron et al., 2015; Plowright et al., 2015).
The ubiquitous occurrence and the vast diversity of bat-associated CoVs have led to the assumption that bats are the primordial hosts of CoVs (Vijaykrishna et al., 2007). Whereas it seems possible that all mammalian CoVs may have originated in bats (Drexler et al., 2014; Woo et al., 2009a, b), this hypothesis should be reevaluated after more complete data on the diversity in other, similarly complex groups of mammalian hosts are available. However, viruses directly infecting human cells have later been found in rhinolophid bats, the primordial natural host of the species SARS-related CoV (Drexler et al., 2014; Ge et al., 2013; Yang et al., 2015).
The investigative work performed on the origins of MERS-CoV suggests that bats are likely the natural reservoirs of this pathogen (Memish et al., 2013). Also camels were implicated in the interspecies transmission of MERS-CoV to humans (Hemida et al., 2014). Multiple studies have shown that there is high seroprevalence of MERS-CoV specific antibodies in dromedary camels (Camelusdromedarius) from Egypt, Oman and other locations in the Middle East, but that sheep, goats, cattle, and other camelids were seronegative (Omrani et al., 2015).
Epidemiological work also showed that a majority of confirmed MERS patients had close contact history with dromedary camels, or products derived from these animals, such as camel meat and milk (Hemida et al., 2017). Furthermore, the MERS-CoV sequences isolated from patients and close-contact dromedary camels were highly similar, or in some cases, 100% identical (Azhar et al., 2014). Thus, the evidence supports that while camels serve as the principal source of MERS-CoV transmission to humans, bats may be again the natural reservoir of this pathogen. (Reusken et al., 2016). Based on the available evidence, it seems that the animal-to-human transmissions of MERS-CoV and SARS-CoV are clear and sets a good paradigm for tracing the origins of HCoV-19.
Therefore, the objectives of this review are: -
2. THE BIOLOGY OF CORONAVIRUS:
2.1. Taxonomy
Coronaviruses (CoVs) are the largest group of viruses belonging to the Nidovirales order, which includes Coronaviridae, Arteriviridae, Mesoniviridae, and Roniviridae families. The Coronavirinae comprises one of two subfamilies in the Coronaviridae family, with the other being the Torovirinae. All viruses in the Nidovirales order are enveloped, non- segmented positive-sense RNA viruses. They all contain very large genomes for RNA viruses, with some viruses having the largest identified RNA genomes, containing up to 33.5 kb genomes (van Regenmortel et al., 2000).
The Coronavirinae are further subdivided into four genera, the alpha, beta, gamma, and delta coronaviruses. The viruses were initially sorted into these genera based on serology but are now divided by phylogenetic clustering (Schoeman and Fielding, 2019).
2.2. Virion structure
Coronavirus virions are spherical with diameters of approximately 125 nm as depicted in recent studies by cryo-electron tomography and cryo-electron microscopy (Neuman et al., 2006). The most prominent feature of coronaviruses is the club-shaped spike projections emanating from the surface of the virion. These spikes are a defining feature of the virion and give them the appearance of a solar corona, prompting the name, coronaviruses. Within the envelope of the virion is the nucleocapsid. Coronaviruses have helically symmetrical nucleocapsids, which is uncommon among positive-sense RNA viruses, but far more common for negative-sense RNA viruses (Schoeman and Fielding, 2019) .
2.3. Genomic organization
As previously mentioned, coronaviruses contain a non-segmented, positive-sense RNA genome of 30 kb. The genome contains a 5′ cap structure along with a 3′ poly (A) tail, allowing it to act as a mRNA for translation of the replicase polyproteins. The replicase gene encoding the nonstructural proteins (Nps) occupies two-thirds of the genome, about 20 kb, as opposed to the structural and accessory proteins, which make up only about 10 kb of the viral genome. The 5′ end of the genome contains a leader sequence and untranslated region (UTR) that contains multiple stem loop structures required for RNA replication and transcription. The 3′UTR also contains RNA structures required for replication and synthesis of viral RNA. The organization of the coronavirus genome is 5′-leader-UTR-replicase- S, E, M, N 3′UTR-poly (A) tail with accessory genes interspersed within the structural genes at the 3′ end of the genome. The accessory proteins are almost exclusively non-essential for replication in tissue culture; however, some have been shown to have important roles in viral pathogenesis (Zhao et al., 2012).

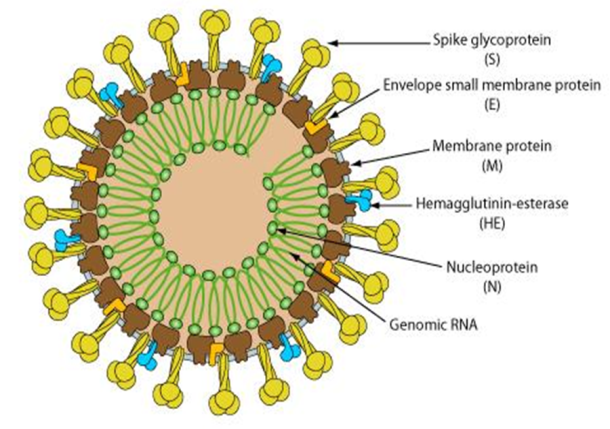
Fig 1. Structural (A) and Genomic (B) organization of α, β, and γ coronavirus of representative.
An illustration of the MHV genome is depicted on top. The expanded regions below show the structural and accessory proteins in the 3’ regions of the MHV, SARS-CoV, and MERS-CoV.
Source: Sciencedirect.com
2.4. Coronavirus life cycle
2.4.1. Attachment and entry
The attachment of the coronavirus to the host cell is mediated by the spike protein and its receptor. The spike protein RBD recognizes and attaches to the ACE2 receptor. Following attachment, the virus can enter the host cell by two different paths. The path the virus takes depends on the host protease available to cleave and activate the receptor-attached spike protein (Simmons et al., 2013).
The first path the coronavirus can take to enter the host cell is by endocytosis and uptake of the virus in an endosome. The receptor-attached spike protein is then activated by the host's pH-dependent cysteine proteasecathepsin L. Activation of the receptor-attached spike protein causes a conformational change, and the subsequent fusion of the viral envelope with the endosomal wall. Alternatively, the virus can enter the host cell directly by proteolytic cleavage of the receptor-attached spike protein by the host's TMPRSS2 or TMPRSS11 Dserine proteases at the cell surface (Simmons et al., 2013).
2.4.2. Genome translation
After fusion the nucleocapsid passes into the cytoplasm, where the viral genome is released. The genome acts as a messenger RNA and the cell's ribosometranslates two-thirds of the genome, which corresponds to the ORF1a and ORF1b, into two large overlapping polyproteins, pp1a and pp1ab. The larger polyprotein pp1ab is a result of a -1 ribosomal frameshift caused by a slippery sequence (UUUAAAC) and a downstream RNA pseudoknot at the end of open reading frame ORF1a. The ribosomal frameshift allows for the continuous translation of ORF1a followed by ORF1b (Fehr and Perlman, 2015).
The polyproteins contain their own proteases, PLpro and 3CLpro, which cleave the polyproteins at different specific sites. The cleavage of polyprotein pp1ab yields 16 nonstructural proteins (nsp1 to nsp16). Product proteins include various replication proteins such as RdRp, RNA helices, and ExoN (Fehr and Perlman, 2015).
2.4.3. Replication and transcription
Model of the replicase-transcriptase complex of a coronavirus. RdRp for replication (red), ExoN for proofreading (dark blue), ExoN cofactor (yellow), RBPs to avoid secondary structure (light blue), RNA sliding clamp for processivity and primasedomain for priming (green/orange), and a helix to unwind RNA (downstream) (Fehr and Perlman, 2015).
A number of the nonstructural replication proteins coalesce to form a multi-protein replicase-transcriptase complex (RTC).The main replicase-transcriptase protein is the RdRp. It is directly involved in the replication and transcription of RNA from an RNA strand. The other nonstructural proteins in the complex assist in the replication and transcription process (Fehr and Perlman, 2015).
The protein nsp15 is a 3'-5' exoribonuclease which provides extra fidelity to the replication process. The exoribonuclease provides a proofreading function to the complex which the RNA-dependent RNA polymerase lacks. Similarly, proteins nsp7 and nsp8 form a hexadecameric sliding clamp as part of the complex which greatly increases the processivity of the RNA-dependent RNA polymerase. The coronaviruses require increased fidelity and processivity during RNA synthesis because of the relatively large genome size in comparison to other RNA viruses (Fehr and Perlman, 2015).
One of the main functions of the replicase-transcriptase complex is to transcribe the viral genome. RdRp directly mediates the synthesis of negative-sense sub-genomic RNA molecules from the positive-sense genomic RNA. This is followed by the transcription of these negative-sense subgenomic RNA molecules to their corresponding positive-sense mRNAs (Fehr and Perlman, 2015). The other important function of the replicase-transcriptase complex is to replicate the viral genome. RdRp directly mediates the synthesis of negative-sense genomic RNA from the positive-sense genomic RNA. This is followed by the replication of positive-sense genomic RNA from the negative-sense genomic RNA (Fehr and Perlman, 2015).
The replicated positive-sense genomic RNA becomes the genome of the progeny viruses. The various smaller mRNAs are transcripts from the last third of the virus genome which follows the reading frames ORF1a and ORF1b. These mRNAs are translated into the four structural proteins (S, E, M, and N) that will become part of the progeny virus particles and also eight other accessory proteins (orf3 to orf9b) which assist the virus (Fehr and Perlman, 2015).
2.4.4. Assembly and release
RNA translation occurs inside the endoplasmic reticulum. The viral structural proteins S, E and M move along the secretary pathway into the Golgi intermediate compartment. There, the M proteins direct most protein-protein interactions required for assembly of viruses following its binding to the nucleocapsid. Progeny viruses are released from the host cell by exocytosis through secretary vesicles (Fehr and Perlman, 2015).
The Coronaviridae are a group of enveloped, positive-strand RNA viruses with non-segmented genomes of about 30,000 nucleotides. The Coronaviridae are further subdivided into four genera, the alpha, beta, gamma, and delta coronaviruses. Which share similarities in the organization and expression of their genomes and the structure of the viral gene products (Siddell, 1995).
3.1. Beta coronavirus
The (β-CoVs or Beta-CoVs) are one of four genera of coronaviruses of the subfamily Orthocoronavirinae in the family Coronaviridae, of the order Nidovirales. They are enveloped, positive-sense, single-strandedRNA viruses of zoonotic origin. The coronavirus genera are each composed of varying viral lineages with the betacoronaviruses genus containing four such lineages (de Groot et al., 2011).
The Beta-CoVs of the greatest clinical importance concerning humans are OC43 and HKU1 of the A lineage, SARS-CoV and SARS-CoV-2 (which causes the disease COVID-19) of the B lineage, and MERS-CoV of the C lineage. MERS-CoV is the first betacoronaviruses belonging to lineage C that is known to infect humans (de Groot et al., 2011).
3.1.1. Linage A: Human Cov -OC43 and HKU1
Lineage A (realm Riboviria, phylum incertaesedis, order Nidovirales, family Coronaviridae, genus Betacoronavirus, subgenus Embecovirus). Embecovirus is a subgenus of coronaviruses in the genus Betacoronavirus. which includes HCoV-OC43 and HCoV-HKU1. The viruses in this subgenus, unlike other coronaviruses, have a HE gene. The viruses in the subgenus were previously known as group 2a coronaviruses (Woo et al., 2007).
The viruses of this subgenus, like other coronaviruses, have a lipid bilayer envelope in which the M, E and S are anchored. Unlike other coronaviruses, viruses in this subgenus also have an additional shorter spike-like structural protein called hemagglutinin esterase (HE) (Woo et al., 2007).
3.1.2. Linage B: SARS-CoV and SARS–Cov2
Lineage B (realm Riboviria, phylum incertaesedis, order Nidovirales, family Coronaviridae, genus Betacoronavirus, subgenus Sarbecovirus) which includes SARSr-CoV (which includes all its strains such as SARS-CoV, SARS-CoV-2, and Bat SL-CoV-WIV1) Sarbecoviruses, unlike embecoviruses or alphacoronaviruses, only have one papain-like proteinase (PLpro) instead of two in the open reading frame ORF1. SARSr-CoV was determined to be an early split-off from the betacoronaviruses based on a set of conserved domains that it shares with the group (Woo et al., 2010).
3.1.3. Linage C: MERS Coronavirus
Lineage C (realm Riboviria, phylum Incertaesedis, order Nidovirales, family Coronaviridae genus Betacoronavirus, subgenus Merbecovirus). Which includes Tylonycteris bat coronavirus HKU4 (BtCoV-HKU4), Pipistrellus bat coronavirus HKU5 (BtCoV-HKU5), and MERS-CoV (various species) Merbecovirus is a subgenus of viruses in the genus Betacoronavirus. The viruses in this subgenus were previously known as group 2 coronaviruses. The viruses of this subgenus, like other coronaviruses, have a lipid bilayer envelope in which the M, E and S structural proteins are anchored (Woo et al., 2007).
The human endemic pathogen HCoV-OC43 based on human tracheal explants kept in organ culture, hence the ‘OC’ in the virus name. The emergence of HCoV-OC43 in humans was proposed to be linked to a host-switching event around the year 1890, It was first isolated in the 1960s a time that coincides with a pandemic of respiratory disease recorded in humans (McIntosh et al., 2005; Vijgen et al., 2005, 2006).
Whether the zoonotic sources for this host transition were cattle, pigs, or other animals are not definitely resolved. However, the high diversity of related Beta CoV1 strains recently described in livestock species support their role as zoonotic sources in the emergence of HCoV-OC43 in humans. Beta CoV1strains are found in many host species is not understood but like other CoVs recombination and deletion events likely played a role in these host-transition or adaptationprocesses. A deletion downstream of the spike gene of HCoV-OC43 was proposed to reflect adaptation to humans Recombination within the species seems to promote the emergence of variants (Vijgen et al., 2005, 2006).
Presence of virus in the central nervous system was reported in patients with chronic demyelinating disease and acute encephalomyelitis (Morfopoulou et al., 2016). The mechanisms that enable neurotropism are not fully understood, but once again mutations within the spike gene may play a role (St-Jean et al., 2004). There is a single study reporting the isolation of BCoV from a 6-year-old child with watery diarrhea (Zhang et al., 1994).
Although the report does not provide information on the patient’s disease and immune status, it reminds us of the potential of BetaCoV1 members to cause spillover infections upon contact with zoonotic sources. Human coronavirus HKU1 (for Hong Kong University) was detected in 2004 in a patient with viral pneumonia (Woo et al., 2005). The virus has subsequently been isolated in tissue cultures and is now recognized as a human respiratory agent. There are nonspecific virus sequences from any other animal species (Pyrc et al., 2010; Woo et al., 2006, 2009b).
However, as HCoV-HKU1 is a sister taxon to MHV and ratsialodacryoadenitis virus, and together with these stands in sister relationship to Beta CoV1 with its basal rodent-associated viruses, a primordial association with rodent hosts may be considered (Wang et al., 2015). Comprehensive studies suggest that the large lineages in the mammalian CoV phylogeny have primordial links to small mammals, focused but not limited to bats (Vijaykrishna et al., 2007; Woo et al., 2009a, b). The enormous effort behind field studies can cause biases in the spectrum of potential host associations studied. Already we sense that rodents, the largest group of mammals, seem to be underrepresented in studies of CoV hosts. The important role of rodents is highlighted by the recent findings of rodent CoVs that hint at primordial host (Guan et al., 2003).
Despite a clear role for intermediate hosts in the transmission of viruses from their natural hosts to humans, we have little information on adaptive processes that contributed by passage through these intermediate hosts (Eckerle et al., 2010; Guan et al., 2003; Wang et al., 2005). Researchers had ideas regarding the emergence of HCoV OC43 that can potentially be refined. It seems peculiar that there is a complete absence of host-specific CoVs in great apes and other primates. This absence provides further support to the suspicion that contact with domestic animals may have been essential in human acquisition of most or all endemic CoV. For HCoV-HKU1 and HCoV- OC43, we therefore should study more of the virus diversity in livestock including closely related. Wildlife species to find footprints of their evolution and emergence. Finally, humans and domestic animals may have carried evolutionary intermediates that explain the emergence of major viral taxa, but that are extinct today (Vijgen et al., 2006)
Discovery of diverse SARS-like coronaviruses in bats several years before the outbreak of SARS, two other zoonotic viruses, Nipah virus and Hendra virus, emerged in Asia and Australia and they were both known to have originated from bats. This led scientists to consider bats in the search of reservoirs of SARS-CoV (Yob et al., 2001). However, subsequent extensive epidemiology studies did not find SARS-CoV in farmed or wild-caught civets, indicating that other animal(s) was involved in SARS-CoV transmission in the animal market or other trading activities and civets are unlikely the natural reservoir of SARS-CoV (Kan et al., 2005).
In 2005, a breakthrough was made as two independent research groups reported, almost simultaneously, the discovery of novel coronaviruses related to SARS-CoV in horseshoe bats (in the genus Rhinolophus) in China, which were termed SARS-like coronavirus (SL-CoV). These bat SL-CoVs from both mainland China eand Hong Kong manifested genome sequence identity of 88–90 % among themselves and 87–92 % identity to human or civet SARS-CoV isolates. The unique set of ORFs exclusively found in SARS-CoV was also present in bat SL-CoVs, demonstrating the close phylogenetic relationship between SARS-CoV and SL-CoV. The discovery of bat SL-CoV boosted researchers’ interest in coronavirus surveillance studies in bats. In the following years, SL-CoV RNA was detected in Rhinolophus species of a wider geographic range in China. The provinces or regions where SL-CoV-positive bats were captured included Hong Kong, Guangxi, Hubei, Shandong, Guizhou, Shaanxi and Yunnan (Ge et al., 2013).
Seven conserved replicase domains in orf1ab of these SL-CoVs found in China were compared with those of SARS-CoV. They all shared higher than 95 % sequence identity with SARS-CoV in the concatenated domains and therefore can be considered to belong to SARS-CoVspecies (King et al., 2012).
In Africa, novel beta coronaviruses related to SARS-CoV have been detected in Hipposideros and Chaerophon species from Ghana, Kenya and Nigeria. However, compared with Asian and European SL-CoVs, these viruses of non-rhinolophid origin were phylogenetically distant to SARS-CoV. The Western African isolates even formed a potential new lineage of Betacoronavirus in the phylogenetic tree (Pfefferle et al., 2009).
On 3rd of January 2020, the first complete genome of the novel β genus coronaviruses (2019-nCoV, subsequently named SARS-CoV-2) was identified in samples of bronchoalveolar lavage fluid from a patient from Wuhan. In recent studies, it has been observed that the novel virus causing epidemics coincides with the CoV isolated in bats. Presence of wild animal trade in Hunan seafood market where the first cases appeared supports this finding. The scientists found that the RBD portion of the SARS-CoV-2 spike proteins had evolved to effectively target a molecular feature on the outside of human cells called ACE2, a receptor involved in regulating blood pressure. The SARS-CoV-2 spike protein was so effective at binding the human cells, in fact, that the scientists concluded it was the result of natural selection and not the product of genetic engineering (Gralinski and Menachery, 2019).
This evidence for natural evolution was supported by data on SARS-CoV-2's backbone and its overall molecular structure. If someone were seeking to engineer a new coronavirus as a pathogen,they would have constructed it from the backbone of a virus known to cause illness. But the scientists found that the SARS-CoV-2 backbone differed substantially from those already known coronaviruses and mostly resembled related viruses found in bats and pangolins (Gralinski and Menachery, 2019).
The amino acid usage pattern of SARS-CoV-2 was generally found like bat and human SARSr-CoVs. He also found greater synonymous codon usage distance between SARS-CoV-2 and its phylogenetic relatives on S and M genes, suggesting these two genes of SARS-CoV-2 are subjected to different evolutionary pressures (Haogao Gu et al., 2020). Based on their genomic sequencing analysis, the most likely origins for SARS-CoV-2 followed one of two possible scenarios. In the first scenario, the virus evolved to its current pathogenic state through natural selection in a non-human host and then jumped to humans. This is how previous coronavirus outbreaks have emerged, with humans contracting the virus after direct exposure to civets (SARS) and camels (MERS). The researchers proposed bats as the most likely reservoir for SARS-CoV-2 as it is very similar to a bat coronavirus. There are no documented cases of direct bat-human transmission, however, suggesting that an intermediate host was likely involved between bats and humans (kristian et al., 2020).
In this scenario, both distinctive features of SARS-CoV-2's spike protein, the RBD portion that binds to cells and the cleavage site that opens the virus up would have evolved to their current state prior to entering humans. In this case, the current epidemic would probably have emerged rapidly as soon as humans were infected, as the virus would have already evolved the features that make it pathogenic and able to spread between people (kristian et al., 2020).
Although the authors found only 4% variability in genomic nucleotides between SARS-CoV-2 and the bat SARSr CoV RaTG13, the difference at neutral sites was 17%, suggesting the divergence between the two viruses is much larger than previously estimated. The report also suggests that new variations in functional sites in the RBD of the spike seen in SARS-CoV-2 and viruses from pangolin SARSr-CoVs are likely caused by mutations and natural selection besides recombination (Xiaolu Tang et al., 2020).
In the other proposed scenario, a non-pathogenic version of the virus jumped from an animal host into humans and then evolved to its current pathogenic state within the human population. For instance, some coronaviruses from pangolins, armadillo-like mammals found in Asia and Africa, have an RBD structure very similar to that of SARS-CoV-2. A coronavirus from a pangolin could possibly have been transmitted to a human, either directly or through an intermediary host such as civets or ferrets (kristian et al., 2020).
Then the other distinct spike protein characteristic of SARS-CoV-2, the cleavage site, could have evolved within a human host, possibly via limited undetected circulation in the human population prior to the beginning of the epidemic. The researchers found that the SARS-CoV-2 cleavage site appears similar to the cleavage sites of strains of bird flu that have been shown to be transmitted easily between people. SARS-CoV-2 could have evolved such a virulent cleavage site in human cells and soon kicked off the current epidemic, as the coronavirus would possibly have become far more capable of spreading between people (kristian et al., 2020).
If the SARS-CoV-2 entered humans in its current pathogenic form from an animal source, it raises the probability of future outbreaks, as the illness-causing strain of the virus could still be circulating in the animal population and might once again jump into humans. The chances are lower of a non-pathogenic coronavirus entering the human population and then evolving properties like SARS-CoV-2 (kristian et al., 2020)
Animal origin of MERS-CoV, SARS-CoV and most early MERS cases had contact history with animals, e.g., dromedary camels (Albarrak et al., 2012). MERS-CoV RNA was detected in camels from Saudi Arabia, Qatar and Egypt and showed high similarities (>99 %) to human MERS-CoV in genomic sequences (Memish et al., 2014). Serological evidence further confirmed a high prevalence of MERS-CoV infections in camels in the Middle East, Africa and Europe (Spain). The neutralization antibodies in camels could be traced back to 1983 (Azhar et al., 2014) The African continent is the last and the least to be affected by COVID 19 pandemic to date (Shabir et al., 2020).
Bat viruses related to MERS-CoV Prior to the emergence of MERS-CoV, a group of bat coronaviruses had been reported including Tylonycteris bat coronavirus HKU4 (BtCoV-HKU4) in Tylonycteris bats and Pipistrellus bat coronavirus HKU5 (BtCoVHKU5) Pipistrellus bats in China (Lau et al., 2014), Eptesicus is abellinus bats in Spain (Falcon et al., 2011) and Pipistrelluspipistrellus bats in the Netherlands (Reusken et al., 2010).
Based on genomic sequence analysis, these bat coronaviruses were grouped into lineage C of the genus Betacoronavirus. After the outbreak of MERS, MERS-CoV related coronaviruses were found in more bat species and countries (Lelli et al., 2013). MERS-CoV derived from camels show high similarities to human MERS-CoV with >99.5 % identities, confirming that the human and camel isolates belong to the same coronavirus species. The most recent ancestor analysis speculated that MERS-CoV may have jumped from bats to camels approximately 20 years ago in Africa, with camels then being imported into the Arabian Peninsula (Corman, et al., 2014).
Although NeoCoV is closer to MERS-CoV than other bat coronaviruses at genomic level, the phylogenetic analysis of the spike protein showed that HKU4 is the most closely related to MERS-CoV among all currently known bat coronaviruses, sharing 67 % sequence identity (Yang et al., 2014). It was suggested that MERS-CoV ancestors had been circulating in bats for a very long time. MERS-CoV has evolved to adapt to use human receptors and the DPP4-recognizing bat coronaviruses like HKU4 may follow up, thereby posing a serious risk to human health (Cui et al., 2013).
Whereas the emergence of SARS involved palm civets, most of the early MERS index cases had contact with dromedary camels. Indeed, MERS- CoV strains isolated from camels were almost identical to those isolated from humans90–95. Moreover, MERS- CoV-specific antibodies were highly prevalent in camels from the Middle East, Africa and Asia13, 14, 96–103. MERS- CoV infections were detected in camel serum samples collected in 1983 (Muller et al., 2014).
Genomic sequence analysis indicated that MERS- CoV, Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5 are phylogenetically related (denoted as betacoronavirus lineage C). The viruses in this lineage have identical genomic structures and are highly conserved in their poly proteins and most structural proteins, but their S proteins and accessory proteins are highly variable. MERSr- CoVs were found in at least 14 bat species from two bat families, Vespertilionidae and Nycteridae. However, none of these MERSr- CoVs is a direct progenitor of MERS- CoV, as their S proteins differ substantially from that of MERS- CoV98 (de Groot et al., 2014).
Overall, all the MERSr- CoVs isolated from bats support the hypothesis that MERS- CoV originated from bats. However, given the phylogenetic gap between the bat MERSr- CoVs and human and camel MERS- CoVs, there should be other yet- to-be- identified viruses that are circulating in nature and directly contributed to the emergence of MERS- CoV in humans and camels. Hopefully, such viruses will be found in bats in the future. However, recombination events have taken place in the evolution and emergence of MERS- CoV94 (Dudas, 2016).
Phylogenetic trees constructed using genes encoding orf1ab and S were incongruent with the tree topology of the complete genome, suggesting potential recombination in these genes 108. Numerous recombinations simply that MERS- CoV originated from the exchange of genetic elements between different viral ancestors, including those isolated from camels and the assumed natural host bats (Luo et al., 2018).
Summary of HCoV: -
|
Type of corona virus |
Intermediate host |
Transmission |
Reported country |
Number of cases |
Death |
|
MERS- CoV |
Camels |
Camels to humans |
Saudi Arabia |
More than 2400 |
774 |
|
SARS-CoV-2. |
Bats |
Bats to human |
Wuhan, Hubei province of china |
77,172,237(Dec,2020) |
1,699,644 (Dec,2020) |
|
SARS |
Bats
|
Bats to civets to human |
Guadong province of china |
8000
|
858
|
|
Rodent |
Rodent to human |
British Columbia |
Not available |
Not available |
|
|
Rodent |
Rodent |
Hong Kong |
585 |
Not available |
STATUS OF CORONA VIRUS IN ETHIOPIA:
Until May 22, 2020, COVID-19 infections were reported from 54 countries in Africa with cumulative confirmed cases of nearly a hundred thousand, and above three thousand deaths. The first confirmed COVID-19 case in this continent was from Egypt and then Algeria. In Ethiopia, the first COVID-19 case was detected and reported on March 13, 2020 (Terefe et al., 2020). Thereafter, in Ethiopia incidences of COVID-19 cases in the country are increasing day to day and as of late May 2020 report, there were a total of 831 confirmed COVID-19 cases and 7 confirmed deaths. Now the coronavirus cases are: 119,951 and 1853 death are reported in December 2020 (WHO, 2020)
In Ethiopia sample from 188 dromedaries, ranging between 1 to 13 years ago were collected as a study evaluating the presence of toxoplasmosis and respiratory tract diseases in 3 provinces including Afar (118 cases), Somalia (11 cases) and Oromia (59 cases) during 2011-2013.All samples were taken by jugular vein puncture according to local laws, and serum samples and were stored at -20 c until they will be tested. All serum samples were shipped to the Erasmus MC laboratory in the Netherlands in agreement with Dutch important regulation. The serum samples were tested for the presence of IgG antibodies reactive with S1 antigens MERS-CoV (CDC, 2014).
As of 5 February 2015, 971 laboratory confirmed cases of human infection with Middle East respiratory syndrome coronavirus (MERS-CoV) have been reported to WHO, including at least 356 deaths. Overall, 63.5% of cases reporting gender n=949 are male and the median age is 48 years (range 9 months -99 years; n=964) (http://www.who.int/csr/disease/coronavirus-infectons/mers-5-february-2015.pdf?ua=1). Despite widespread seropositivity for MERS-CoV reported in dromedaries throughout the Middle East, North Africa and east Africa, human MERS cases have been observed only in the Middle East with no human case reported in Africa to date (Liljander et al., 2020)
Conclusions Although the study of bat-borne coronaviruses has only started just about 10 years ago, the scientific community has already learnt a great deal of useful lessons which will be instrumental in mitigating, predicting, and preventing future zoonotic coronavirus outbreaks. Bats harbor coronaviruses with great genetic diversity. It is believed that most, if not all, currently circulating alpha coronaviruses and beta coronaviruses in different mammals are evolutionarily linked to ancestral coronaviruses originating from bats. Different species of rhinolophid bats in China carry genetically diverse SARS-like coronaviruses, some of which are direct ancestors of SARS-CoV and hence have the potential to cause direct interspecies transmission to humans. Meanwhile, different coronavirus species closely related to MERS-CoV are circulating in bats.
Based on the above conclusion, the following recommendations are forwarded.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org