International Journal of Epidemiology And Public Health Research
OPEN ACCESS | Volume 9 - Issue 1 - 2026
ISSN No: 2836-2810 | Journal DOI: 10.61148/2836-2810/IJEPHR


Nan Lu*, Junling Duan
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
*Corresponding author: Nan Lu, College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
Received: April 01, 2025 Accepted: April 10, 2025 Published: April 15, 2025
Citation: Nan Lu, Junling Duan. (2025) “College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China”. International Journal of Epidemiology and Public Health Research, 6(2); DOI: 10.61148/2836- 2810/IJEPHR/122.
Copyright: © 2025. Nan Lu. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited., provided the original work is properly cited.
The first theoretical investigation was provided by our DFT calculation on Tf2O-catalyzed [3 + 2 + 1] benzannulation of annular-enaminone with monomeric benzoyl acetonitrile. Initially activated by Tf2O, enaminone was formed vinyl iminium triflate intermediate, which was captured by benzoyl acetonitrile to initiate selective 1,2-addition and form first TfOH. Subsequently, key trifluorome-thanesulfonate intermediate was generated after elimination of dimethylamine. Next with a second molecule of benzoyl acetonitrile, the reaction took place via Michael-type addition followed by second proton donation. Then competitive 1,4-elimination or 1,2-elimination were possible to complete the release of second TfOH. Finally, the product multisubstituted arylnitrile was obtained through sequential Knoevenagel condensation and aromatization. Michael-type addition was determinate to be rate-limiting from the perspective of whole process. The 1,2-elimination was more favorable within two parallel paths from kinetics.
1 Introduction
As common functional group, CN is easily converted into aldehydes and amidines. This makes aromatic nitriles versatile organic synthons for a wide range of natural products and agricultural chemicals [1,2]. Many cross-coupling reactions were efficient to synthesize aromatic nitriles [3-5]. In recent years, many efforts have been focused on benzannulation offering a powerful strategy for constructing aromatic rings from acyclic precursors. Du reported Copper-catalyzed [3 + 2 + 1] cycloaddition of alkenes with benzoquinones and dicarbonyl compounds [6]. Chen explored [3 + 3] cycloaddition reaction of vinyl sulfoxonnium ylides with cyclopropenones [7]. Wei researched [2 + 2 + 1 + 1] cycloaddition for de novo synthesis of densely functionalized phenols [8]. There are also base-mediated benzannulation of α-cyanocrotonates with ynones and five-component [2 + 2 + 1 + 1] tandem benzannulation to multifunctionalized aromatic amines [9,10].
A versatile class of push−pull olefin we are interested in is enaminone as promising precursor for various aromaticcompounds [11,12]. For instance, Zeng discovered photocatalytic pyridine synthesis with enaminones and TMEDA under metal-free conditions [13]. Fan researched Co(III)-catalyzed coupling of enaminones with oxadiazolones for imidazole synthesis [14]. Zhang reported Silver-catalyzed cascade bis-heteroannulation reaction of enynones and o-hydroxyphenyl enaminones to access highly functionalized 3-furylmethyl chromones [15]. Duan gave chemodivergent synthesis of polysubstituted pyrroles and pyridines via tandem site-selective bromination and highly regioselective Heck reaction of N-allyl enaminones [16]. Usually, enaminone participates in benzannulation as Cn synthon. Such as redox neutral [4 + 2] benzannulation of dienals and tertiary enaminones in benzaldehyde synthesis, Copper(II)-catalyzed [2 + 2+2] annulation of enaminones with maleimides using traceless directing group, [3 + 3] condensation reaction from tricarbonyl compounds and enaminones, cinnamaldehydes for synthesis of polysubstituted phenols, and [5 + 1] cyclizations via cleavage of 1,3-dicarbonyls to synthesize highly functionalized naphthols [17-20]. However, [3+ 2 + 1] benzannulation remained less.
In this field, trifluoromethanesulfonic anhydride (Tf2O) is an efficient activation reagent for carbonyl derivatives [21]. Zhang group has made many contributions in converting enaminones into vinyl iminium triflates. The previous work includes one-pot synthesis of 3-amino diynes via Cu(I)-catalyzed reaction with terminal alkynes, Tf2O-mediated tandem reaction to functionalized conjugated-enals/β-naphthalaldehydes, and divergent synthesis of enynals and dihydrobenzo[f]isoquinolines [22-24]. Another breakthrough was Tf2O‑mediated [3 + 2 + 1] benzannulation of enaminones with acylacetonitriles [25]. Although multisubstituted arylnitriles were synthesized, many problems in experiment still puzzled requiring an in-depth theoretical study for this strategy. How annular-enaminone was activated by Tf2O to form vinyl iminium triflate? How crucial trifluorome-thanesulfonate intermediate was generated during elimination of dimethylamine? What’s the detailed process of sequential Knoevenagel condensation and aromatization following either 1,4- or 1,2-elimination?
2 Computational details
Structures were optimized at M06-2X/6-31G(d) level with GAUSSIAN09 [26]. Among various DFT methods [27], M06-2X functional has smaller deviation between experimental and calculated value than B3LYP hybrid functional [28,29]. With 6-31G(d) basis set, it can provide best compromise between time consumption and energy accuracy. It was also found to give accurate results for stepwise (2 + 2) cycloaddition, enantioselective (4 + 3) and Diels−Alder reaction [30,31]. Together with good performance on noncovalent interaction, it is suitable for this system [32-34]. To obtain zero-point vibrational energy (ZPVE), harmonic frequency calculations were carried out at M06-2X/6-31G(d) level gaining thermodynamic corrections at 353 K and 1 atm in dichloromethane (DCM). At M06-2X/6-311++G(d,p) level, the solvation-corrected free energies were obtained using integral equation formalism polarizable continuum model (IEFPCM) [35-39] on M06-2X/6-31G(d)-optimized geometries. NBO procedure was performed with Natural bond orbital (NBO3.1) obtaining lone pair and bond to characterize bonding orbital interaction and electronic properties [40-42]. Using Multiwfn_3.7_dev package [43].
3 Results and Discussion
The mechanism was explored for Tf2O-catalyzed [3 + 2 + 1] benzannulation of annular-enaminone 1 with monomeric benzoylacetonitrile 2 to multisubstituted arylnitrile 3 (Scheme 1). Illustrated by black arrow of Scheme 2, under the activation of Tf2O, enaminone 1 formed vinyl iminium triflate intermediate A, which was subsequently captured by benzoyl acetonitrile 2 to initiate selective 1,2-addition forming intermediate B and TfOH. Subsequently, key trifluorome-thanesulfonate intermediate 4 was generated after elimination of dimethylamine from B. Then Michael-type addition of 4 reacted with a second molecule of 2 gave intermediate C followed by second proton donation from 2. The intermediates D or E were yielded via competitive 1,4-elimination (red arrow) or 1,2-elimination (blue arrow) after the release of a second TfOH. Finally, sequential Knoevenagel condensation and aromatization occurred resulting in final product multisubstituted arylnitrile (D to F to 3, or E to G to 3). Figure 1 listed schematic structures of optimized TSs in Scheme 2. Table 1 gave activation energy for all steps.
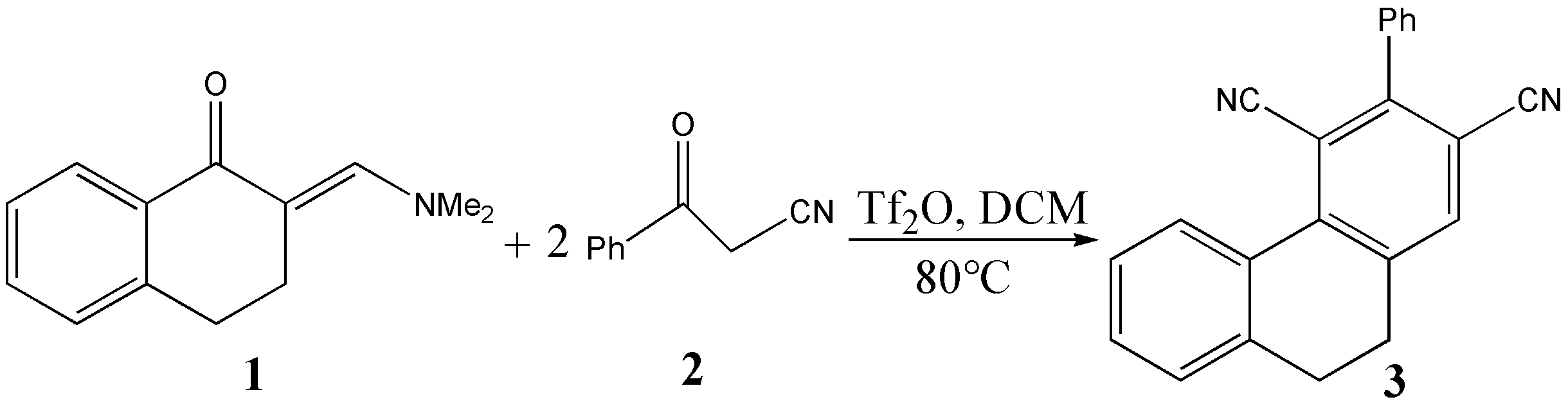
Scheme 1 Tf2O-catalyzed [3 + 2 + 1] benzannulation of annular-enaminone 1 with monomeric benzoylacetonitrile 2 to access multisubstituted arylnitrile 3.
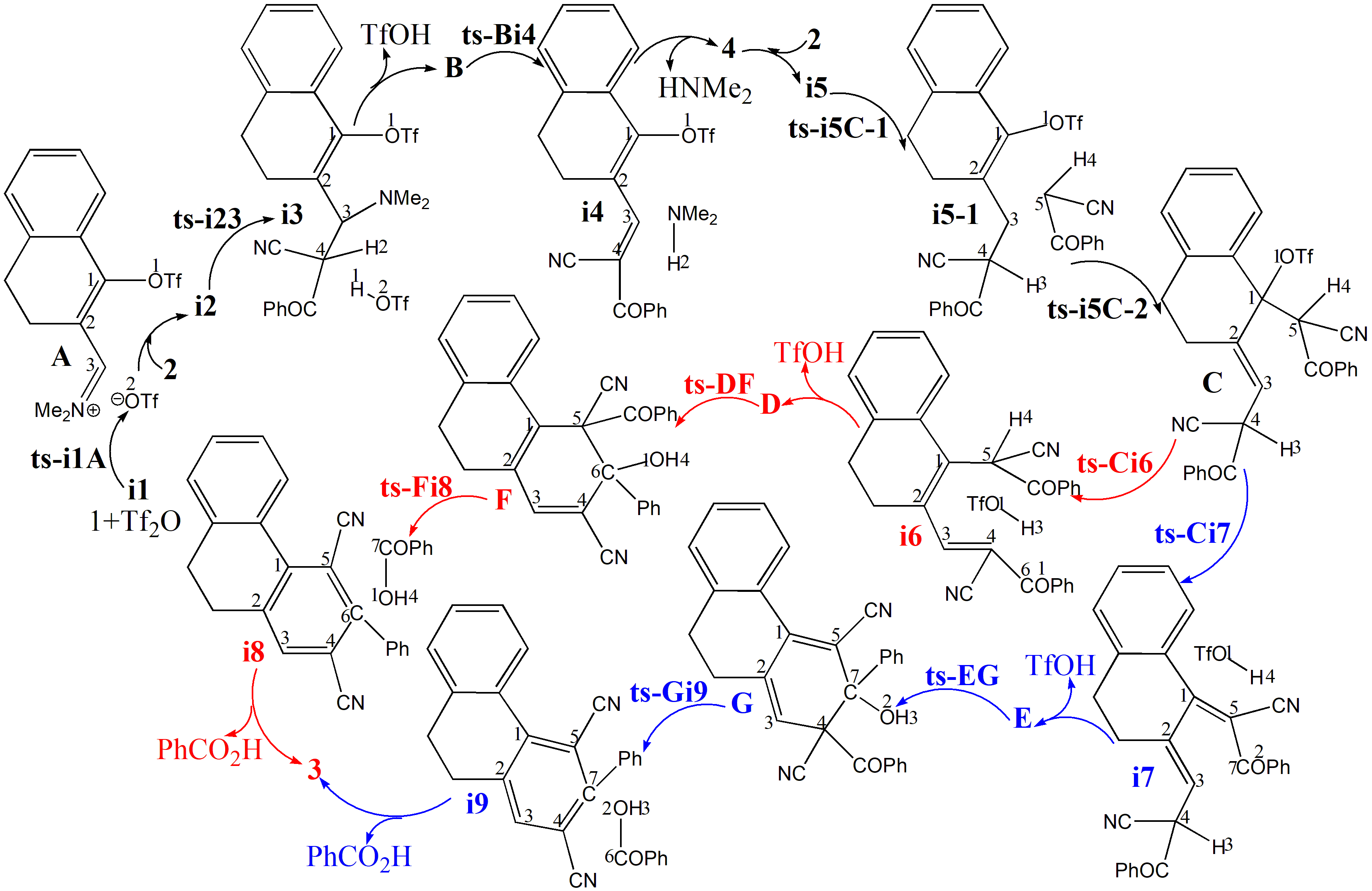
Scheme 2 Reaction mechanism of Tf2O-catalyzed [3 + 2 + 1] benzannulation of 1 with 2 to access 3.
3.1 Vinyl iminium triflate formation/selective 1,2-addition/dimethylamine elimination
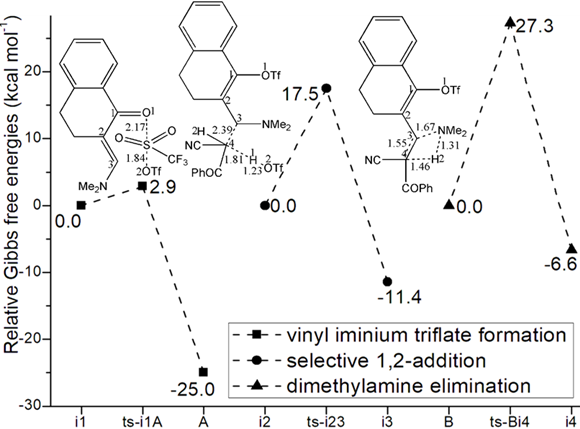
Initially from complex i1 binding enaminone 1 and catslyst Tf2O, vinyl iminium triflate intermediate A was formed via ts-i1A in step 1 with low activation energy of 2.9 kcal mol−1 exothermic by -25.0 kcal mol−1 (black dash line of Figure 1a). The transition vector includes one Tf leaving from Tf2O bonding to carbonyl of 1 that is O2···S···O1 (1.84, 2.17 Å). The activation of Tf2O is revealed through small barrier and the resultant A rather stable.
Subsequently, the intermediate i2 was taken as new starting point afte A captured by benzoyl acetonitrile 2. Thus selective 1,2-addition was initiated via ts-i23 in step 2 with activation energy of 17.5 kcal mol−1 exothermic by -11.4 kcal mol−1 generating i3. Prior to the nucleophilic addition of C4 of 2 to C3 of 1, the transition vector also demonstrates concerted proton transfer C4···H1···O2 (2.39, 1.81, 1.23 Å). Once C3-C4 typical single bond was formed, the first molucule of TfO2H1 was also obtained. After the removal of TfOH, stable intermediate B was in hand for next step.
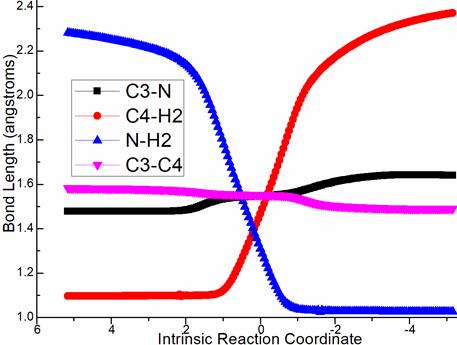
Via ts-Bi4, dimethylamine elimination occurs in step 3 with activation energy of 27.3 kcal mol−1 exothermic by -6.6 kcal mol−1 delivering intermediate i4. The transition vector corresponds to dissociation of NMe2 from C3, H2 from C4 and the resulting linkage of H···NMe2 as HNMe2 molecule, strengthened C3=C4 new double bond (1.67, 1.46, 1.31, 1.55 Å) (Figure S1a). When dimethylamine HNMe2 was eliminated, the key trifluorome-thanesulfonate intermediate was generated denoted as 4 involving stable conjugated diene structure.
Table 1 The activation energy (in kcal mol−1) of all reactions in gas and solvent
|
TS |
ΔG≠gas |
ΔG≠sol |
|
ts-i1A |
4.6 |
2.9 |
|
ts-i23 |
22.6 |
17.5 |
|
ts-Bi4 |
30.2 |
27.3 |
|
ts-i5C-1 |
35.1 |
29.3 |
|
ts-i5C-2 |
32.6 |
27.6 |
|
ts-Ci6 |
19.9 |
17.4 |
|
ts-Ci7 |
19.4 |
16.6 |
|
ts-DF |
38.0 |
31.9 |
|
ts-Fi8 |
29.9 |
20.1 |
|
ts-EG |
29.8 |
27.3 |
|
ts-Gi9 |
19.6 |
14.8 |
(a)
(b)
(c)
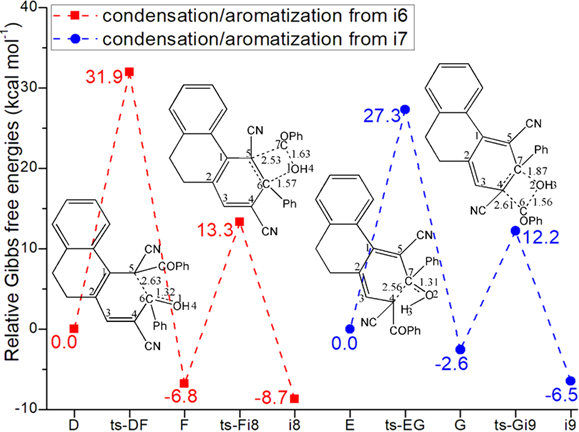
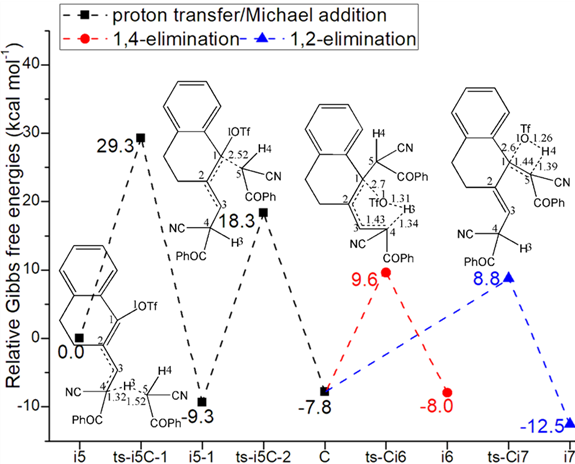
Fig. 1 Relative Gibbs free energy profile in solvent phase starting from complex (a) i1, i2, B (b) i5 (c) D, E (Bond lengths of optimized TSs in Å).
3.2 Proton transfer/Michael addition/1,4- or 1,2-elimination
With a second molecule of 2 added to 4, i5 was in hand as new starting point of next three steps (black dash line of Figure 1b). In step 4, C4 is protonated via ts-i5C-1 with activation energy of 29.3 kcal mol−1 exothermic by -9.3 kcal mol−1 delivering intermediate i5-1. The transition vector corresponds to proton donation from 2 C5···H3···C4 (1.52, 1.32 Å). This not only makes C4 sp3 hybrid but enhances the nucleophilic ability of C5 ready for the following Michael-type addition. With decreased activation energy of 27.6 kcal mol−1, the nucleophilic attack C5···C1 (2.52 Å) happens via ts-i5C-2 in step 5 exothermic by -7.8 kcal mol−1 giving intermediate C. The double bond turns to be located on C2=C3 in C.
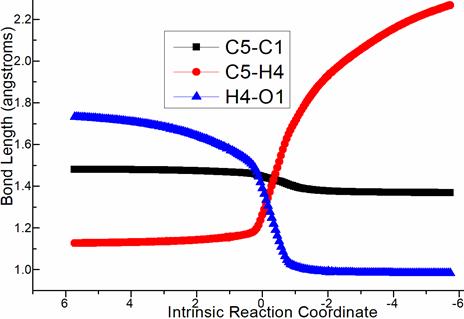
Then competitive 1,4- or 1,2-elimination (red, blue dash line of Figure 1b) were possible for step 6. The common point is cleavage of TfO1 from C1 via C1···O1 breaking. The difference lies in through ts-Ci6, H3 is departed from C4 to O1 as TfO1H3 while H4 is disruptured from C5 to O1 as TfO1H4 via ts-Ci7. The activation energy is 17.4, 16.6 kcal mol−1 exothermic by -8.0, -12.5 kcal mol−1 generating i6, i7 respectively. The transition vector is also similar with the case of ts-Ci7 including C1···O1 and C5···H4···O1 (2.6, 1.39, 1.26 Å) (Figure S1b). After the release of a second TfOH molecule from i6 or i7, the intermediates D or E were yielded with recovered conjugated diene structure of 4 yet different position.
3.3 Parallel Knoevenagel condensation and aromatization
The sequential Knoevenagel condensation and aromatization occurred from D or E in parallel mode both leading to final product multisubstituted arylnitrile 3. Via ts-DF in step 7, the activation energy of 31.9 kcal mol−1 exothermic by -6.8 kcal mol−1 affording ring closure of F (red dash line of Figure 1c). The transition vector includes closure of six membered ring via C5···C6 bonding and cooperative stretching of C6···O1 from double to single (2.63, 1.32 Å). Besides novel hydroxyl O1H4, typical C5-C6 single bond is available in F, from which the aromatization proceeds via ts-Fi8 in step 8 with activation energy is 20.1 kcal mol−1 exothermic by -8.7 kcal mol−1 generating i8. The transition vector contains breaking of C5···C7, C6···O1 as well as C7···O1 connecting as PhCOOH molecule and contracted C5-C6 from single to double completing aromatization.
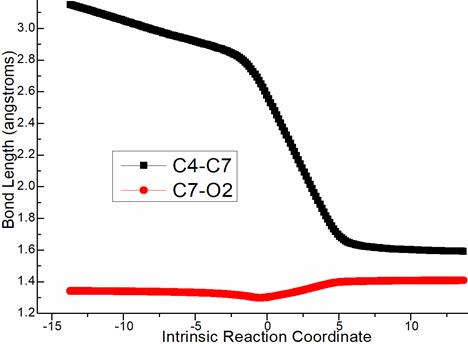
Alternatively via ts-EG in step 7, the activation energy is reduced to be 27.3 kcal mol−1 exothermic by -2.6 kcal mol−1 realizing G (blue dash line of Figure 1c). The transition vector indicates nucleophilic approach of C4···C7 (2.24 Å) and C7···O2 stretching from double to single (2.56, 1.31 Å) (Figure S1c). The structure of G is characterized by hydroxyl O2H3 and C4-C7 single bond ready for aromatization via ts-Gi9 in step 8 with activation energy of 14.8 kcal mol−1 exothermic by -6.5 kcal mol−1 generating i9. The transition vector includes PhCO, O2H3 breaking via C4···C6, C7···O2 and PhCOOH molecule formation via C6···O2 connecting, aromatization via C4=C7 contraction from single to double (2.61, 1.87, 1.56 Å) (Figure S1d). From the perspective of whole process, Michael-type addition was determinated to be rate-limiting step. The 1,2-elimination was more favorable within two parallel paths kinetically.
Table S1. Calculated relative energies (all in kcal mol-1, relative to isolated species) for the ZPE-corrected Gibbs free energies (ΔGgas), Gibbs free energies for all species in solution phase (ΔGsol) at 353 K by M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method and difference between absolute energy.
|
Species |
ΔGgas |
ΔGsol(EtOH) |
|
1+Tf2O |
0.00 |
0.00 |
|
i1 |
-11.14 |
-6.51 |
|
ts-i1A |
-6.59 |
-3.66 |
|
A |
-19.85 |
-31.50 |
|
A+2 |
0.00 |
0.00 |
|
i2 |
-2.50 |
-3.85 |
|
ts-i23 |
20.13 |
13.64 |
|
i3 |
-12.63 |
-15.26 |
|
i3-TfOH |
0.00 |
0.00 |
|
B |
26.89 |
26.43 |
|
ts-Bi4 |
57.08 |
53.68 |
|
i4 |
20.73 |
19.81 |
|
i4-HNMe2 |
0.00 |
0.00 |
|
4 |
-3.58 |
-4.86 |
|
4+2 |
0.00 |
0.00 |
|
i5 |
-4.85 |
-8.58 |
|
ts-i5C-1 |
30.21 |
20.71 |
|
i5-1 |
-15.72 |
-17.92 |
|
ts-i5C-2 |
16.87 |
9.76 |
|
C |
-10.43 |
-16.37 |
|
ts-Ci6 |
9.46 |
1.01 |
|
i6 |
-10.49 |
-16.54 |
|
ts-Ci7 |
9.02 |
0.20 |
|
i7 |
-13.99 |
-21.11 |
|
i6-TfOH |
0.00 |
0.00 |
|
D |
2.63 |
-0.87 |
|
ts-DF |
40.62 |
31.07 |
|
F |
-6.03 |
-7.71 |
|
ts-Fi8 |
23.86 |
12.41 |
|
i8 |
-6.84 |
-9.60 |
|
i8-PhCOOH |
0.00 |
0.00 |
|
3 |
-29.13 |
-25.67 |
|
i7-TfOH |
0.00 |
0.00 |
|
E |
-5.27 |
-8.08 |
|
ts-EG |
24.56 |
19.23 |
|
G |
-6.15 |
-10.67 |
|
ts-Gi9 |
13.41 |
4.14 |
|
i9 |
-10.78 |
-14.56 |
Table S2. The activation energy (local barrier) (in kcal mol−1) of all reactions in the gas, solution phase calculated with M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method.
|
TS |
ΔG≠gas |
ΔG≠sol |
|
ts-i1A (134i) |
4.6 |
2.9 |
|
ts-i23 (112i) |
22.6 |
17.5 |
|
ts-Bi4 (1596i) |
30.2 |
27.3 |
|
ts-i5C-1 (1091i) |
35.1 |
29.3 |
|
ts-i5C-2 (346i) |
32.6 |
27.6 |
|
ts-Ci6 (1182i) |
19.9 |
17.4 |
|
ts-Ci7 (996i) |
19.4 |
16.6 |
|
ts-DF (287i) |
38.0 |
31.9 |
|
ts-Fi8 (177i) |
29.9 |
20.1 |
|
ts-EG (396i) |
29.8 |
27.3 |
|
ts-Gi9 (189i) |
19.6 |
14.8 |
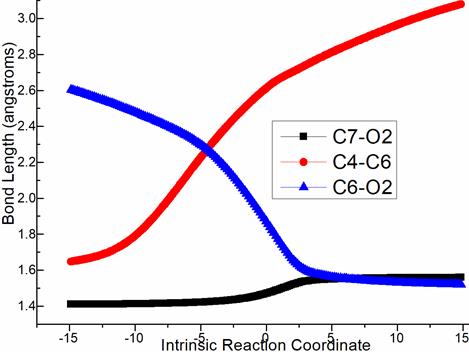
Figure S1. Evolution of bond lengths along the IRC for (a) ts-Bi4 (b) ts-Ci7 (c) ts-EG (c) ts-Gi9 at M06-2X/6-311++G(d,p) level.
(a) (b)
(c) (d)
4 Conclusions
The first theoretical investigation was provided by our DFT calculation on Tf2O-catalyzed [3 + 2 + 1] benzannulation of annular-enaminone with monomeric benzoyl acetonitrile. Activated by Tf2O, enaminone was initially formed vinyl iminium triflate intermediate, which was captured by benzoyl acetonitrile to initiate selective 1,2-addition and form first TfOH. Next, key trifluorome-thanesulfonate intermediate was generated after elimination of dimethylamine. Subsequently with a second molecule of benzoyl acetonitrile, the reaction took place via Michael-type addition followed by second proton donation. Then competitive 1,4-elimination or 1,2-elimination were possible to complete the release of second TfOH. Finally, the product multisubstituted arylnitrile was obtained through sequential Knoevenagel condensation and aromatization. Michael-type addition was determinated to be rate-limiting from the perspective of whole process. The 1,2-elimination was more favorable within two parallel paths kinetically.
Electronic Supplementary Material
Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 353 K.]
Author contributions: Conceptualization, Nan Lu; Methodology, Nan Lu; Software, Nan Lu; Validation, Nan Lu; Formal Analysis, Nan Lu; Investigation, Nan Lu; Resources, Nan Lu; Data Curation, Nan Lu; Writing-Original Draft Preparation, Nan Lu; Writing-Review & Editing, Nan Lu; Visualization, Nan Lu; Supervision, Nan Lu; Project Administration, Nan Lu; Funding Acquisition, Junling Duan. All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, P.R. China.
Conflict of interest: The authors declare no conflict of interest.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org