Pharmacy and Drug Innovations
OPEN ACCESS | Volume 4 - Issue 1 - 2025
ISSN No: 2994-7022 | Journal DOI: 10.61148/2994-7022/PDI

Hindustan Abdul Ahad*, Haranath Chinthaginjala, Pasupuleti Dheeraj Krishna, Gangireddy Jayasimha Reddy, Syed Rahamathulla
Department of Industrial Pharmacy, Raghavendra Institute of Pharmaceutical Education and Research (RIPER) - Autonomous, Ananthapuramu-515721, AP, India.
*Corresponding author: Hindustan Abdul Ahad, Department of Industrial Pharmacy, Raghavendra Institute of Pharmaceutical Education and Research (RIPER) - Autonomous, Ananthapuramu-515721, AP, India.
Received: June 06, 2021
Accepted: June 16, 2021
Published: June 18, 2021
Citation: Hindustan Abdul Ahad, Haranath Chinthaginjala, Pasupuleti Dheeraj Krishna, Gangireddy Jayasimha Reddy, Syed Rahamathulla. “Investigational New Drug the Leaping Step Before Committing Trials in Humans: An Informative Note”. J Pharmacy and Drug Innovations, 2(4); DOI: http;//doi.org/03.2020/1.1020.
Copyright: © 2021 Hindustan Abdul Ahad. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Clinical examiners summon a few explicit administrative prerequisites if their examination incorporates the utilization of a drug specialist. Studies using a medication that the Food has not endorsed and Drug Administration (FDA) or for indications not in the supported marking may require recording an Investigational New Drug (IND) application with the FDA. On the off chance that an investigation meets explicit administrative exclusion models, an IND may not be required. Individual specialists may meet the FDA meaning of a support examiner, in which case the application interaction is mostly less muddled than for business backers, and this audit tends to just the present situation. Recording an IND requires finish of 3 arrangements of structures: 1 itemizing the examination (FDA Form 1571), 1 giving data about the examiner and study site (FDA Form 1572), and 1 ensuring that the investigation is enlisted in the public data set of clinical preliminaries (FDA Form 3674). If the IND is endorsed, the investigation may start 30 days after the FDA perceives receipt and doles out an IND. On the off chance that the FDA requires extra data or if the examination is put on a "clinical hold," the investigation should not proceed. While the IND is dynamic, the examiner should likewise keep on gathering a bunch of guidelines for checking the examination and answering to the FDA.
Introduction
Clinical inspectors creating drug studies call upon several detailed regulatory needs further than those mandates for the defence of human subjects in clinical trials (Petryna, 2009; Postal & Diaz, 2011). These regulatory needs for medicine studies deal with the protection and effectiveness issues exclusive to the make use of pharmaceuticals in the clinical examination background. The U.S. Food and Drug Administration (FDA) is exciting with the guideline of most drugs besides previous goods (Marchetti & Schellens, 2007). This extends to regulatory authority above clinical study via these agents. As a result, to carry out medical studies, a researcher has to obey FDA’s needs. Deteriorating to meet up the FDA’s rules can cover official and economic implications intended for the persons performing the investigation besides the institutions connected among the research manners (Jin, et al., 2017).
A primary division of regulatory procedure implicated for trial drugs is notifying the FDA that a pharmaceutical agent resolve subsists previous within an experimental way. This announcement is called the Investigational New Drug (IND) application (Kelly et al., 2014). In favour of medicine trials performed by the pharmaceutical industry or other business sponsors, persons greatly skilled and professional in gathering the rules deal with the regulatory needs. Though, for character investigators who are not as well-known with the needs and rules, file an IND can be unapproachable and may be supposed as an obstruction to performing drug studies. It is attractive to a reminder that the common IND submissions are non-commercial (Hecht et al., 2014). Therefore, human being clinical investigators often gather the regulatory desires essential to perform investigational medicine studies. This re-evaluation is anticipated to deal with the simplest situation in which an individual researcher starts and performs a drug study that requires filing and continues an IND among FDA. In addition, intended for a need of straightforwardness, the aforementioned check just addresses regulatory desires for studies performed at a particular site. Figure (1) represents the IND function procedure used for a sponsor-investigator (Jha et al., 2021).
This purpose regarding direction towards helping the sponsor-investigators over the production and put forward the entire investigational new drug applications (INDs) towards the Centre for Drug Evaluation and Research (CDER) and the Centre for Biologics Evaluation and Research (CBER) at the Food and Drug Administration (FDA) (Holbein, 2009). Sponsor-investigators looking forward to conducting clinical researches often do not? Include this regulatory awareness otherwise this capital towards appointed professionals headed for helping sponsor-investigator by this IND giving in the procedure. Even though neither the complete bit by bit training guidebook, the regulation calls attention to a definite fundamental about the procedure towards assist sponsor-investigators doing well compliance about IND (Lapteva & Pariser, 2016). The control in addition discusses the IND revaluation method and also common tasks about sponsor-investigators associated with the clinical investigation. This is significant towards reminder with the aim of the regulation will not comprise negotiations about everyone about these needs to affect towards this IND compliance also re-evaluate method or else towards performing the clinical study. Sponsor-investigators have to re-examine within complete their requests, which describes during this Code of Federal Regulations (CFR). A lot of sections about these systems with this purpose about influence toward INDs are reported otherwise hinted just before within the supervision. Details about this educational substance about IND with details required towards total essential shape too are present all over the regulation (Thakur et al., 2017).
The regulation is engaged foremost by individual sponsor-investigators looking towards estimate medicine to be both presently accepted otherwise been examined below an active IND intended for the dissimilar suggestion (Chiodin et al., 2019). The management is designed for sponsor-investigators for just beginning the medicine designed for-profit and therefore do not centre of attention taking place on definite regulatory desires to engage replace about particulars or else resources linking sponsor and investigator. The management will not relate to clinical trials to facilitate any requirement towards conducting beneath IND. The management too is not projected towards a deal with prolonged contact INDs otherwise biologic devices. Sponsor-investigators have to consign towards obtainable FDA set of laws and regulation and/or get in touch by this applicable CDER or CBER re-examine partition towards converse and gain extra particulars meant for producing INDs which is not covered with the regulation (Jarow et al., 2016).
FDA’s regulation pass will not start officially enforceable responsibilities. Instead, guidance’s described this department’s modern opinion lying on a theme and must be analysis no more than a proposal, except definite regulatory otherwise legislative wants be quiet. This applies about this phrase must be within organization guidance funds with the purpose of somewhat be optional otherwise suggested, however not mandatory (Shapiro, 2002).
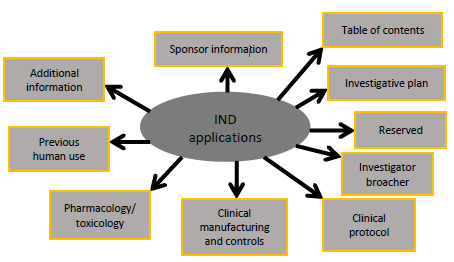
Fig.1: Applications of IND
Normally the FDA set of laws involve sponsors, and sponsor-investigators, who desire towards calculating a medicine otherwise biological creation within human being on the way to give in IND towards this FDA (21 CFR part 312). This FDA’s most important intention during re-evaluations of the IND is to help shield these privilege and protection about themes and, within phases 2 along with 3, to assist make sure of this quality about this clinical trial is been satisfactory towards estimate this drug’s efficacy and safety (Wonnacott et al., 2008).
The sponsor takes duty meant for and starts the clinical investigation. Sponsor be capable of subsisting an entity otherwise pharmaceutical business, governmental organization, intellectual institution, private association, otherwise previous association. An investigator is a character who performs this investigation (Holbein et al., 2014).
Sponsor-investigator is one who equally begins and performs research, and below instant route this investigational medicine does admin otherwise dispensed. This phrase, while definite within the FDA set of laws; Does not contain several entities previous to individual. At the same time as the name recommend, sponsor-investigator presupposes this task and has to obey by FDA set of laws related to together a sponsor along with an investigator. These tasks comprise this giving in with protection about IND (Van, 2018).
A sponsor-investigator may not be required toward giving in IND for example, a study of officially marketed medicine but these criterions intended for IND exception be meet. During various conditions, still but sponsor-investigator be a mandatory route for giving in an IND, this IND will not require including everyone about this planned over. For example, about sponsor-investigator be tendering towards estimating medicine with the intention of this subject about an active IND, sponsor-investigator can seek a note about annotation agreement beginning this sponsor about with the purpose of IND (called the commercial sponsor) that permit this sponsor-investigator on the way to consign this FDA toward the particulars enclosed within this trade sponsor’s IND. But this sponsor-investigator be evaluating FDA-approved prescription or non-prescription drugs, even if an IND is required, some of the information needed for an IND submission can be found in the FDA-approved labelling (Perez et al., 2016). The flow diagram involve in the IND submission is illustrated in figure 2.

Fig. 2: Flow diagram designed for clinical medicine evaluation to involve in IND submission intended for investigator-sponsor
Regulatory situation and FDA responsibility
Food and Drug Administration (FDA) exist as an organization within the United States division of wellbeing and Human forces stimulating with assuring safety and effectiveness about human with veterinary drugs besides the previous region of regulatory authority. The organization also dependable intended to make possible progress within medications. This FDA association is the large and somewhat complicated essential association by quantity about centres, divisions, and agencies located commonly centrally within the Washington Metropolitan Area additionally on the way to a range of locality organization within the United States. Intended for this cause about regulatory have power over about investigational medicines within clinical trials that be conducted on the human being, this organization mostly occupied are the Centre for Drug Evaluation and Research (CDER), Centre for Devices and Radiological Health, and the Centre for Biologics Evaluation and Research (CBER). Above organizations are offices among regulatory, purposeful, otherwise therapeutic focus. Pharmaceutical products will be both man-made and natural, fall beneath this regulatory direction about CDER, and most drug studies (Vu et al., 2015). The CBER makes confirms biological and related products including blood, vaccines, allergens, tissues, and cellular and gene therapies. As result, only a little number of focused drug studies would appear in CBER jurisdiction. FDA Web site publishes complete directorial charts through the names and contact information of officials.
The prime set of central laws establishing FDA power and a codification of the rules is Federal Food, Drug, and Cosmetic Act. The exact part about the mentioned laws covering an IND is in Part 312 of the Code of Federal Regulations (CFR). Close at hand are too preceding sections of this CFR which contact the behaviour about clinical evaluations by utilising pharmaceutical products. Table 1 mentions extra major sections appropriate on the way to individual investigators. Every one of sets of about sections is gladly reachable on the FDA Web site within a searchable system (McElvany, 2009). Finally, Federal law order with the purpose of demand designed for medicine, which is transported otherwise scattered across position appearance, which must have accepted selling request. Because medicines which are to be utilised within all clinical trials are elated through different state lines, this sponsor is obliged to look for let go with the aim of formally certified obligation. This with the name of, “Take in regarding claim Investigational exception used for New Drug” refers to exception. This commonly used to mention this is IND.
Code for federal Regulations (Herwaldt et al., 2018).
|
21 CFR Part 312 |
IND application |
|
312.2 |
Applicability |
|
312.23 |
IND content and format |
|
312.30 |
Protocol amendments |
|
312.31 |
Information amendments |
|
312.32 |
IND safety reports |
|
312.33 |
Annual reports |
|
312.38 |
Withdrawal of an IND |
|
312.42 |
Clinical holds and requests meant for alteration. |
|
312.44 |
Termination |
|
312.45 |
Inactive status |
|
312.50 |
Responsibilities of sponsors |
|
312.60 |
General responsibilities of Investigators |
|
312.61 |
Control about the investigational drug |
|
312.62 |
Investigator recordkeeping and record retention |
|
312.64 |
Investigator reports |
|
312.66 |
Assurance of IRB review |
|
312.68 |
Inspection about investigator’s records and reports |
|
312.69 |
Handling of controlled substances |
|
312.70 |
Disqualification about a clinical investigator |
|
Other relevant regulations |
|
|
21 CFR Part 314 |
IND, and NDA purpose, meant for FDA consent to market a new drug |
|
21 CFR Part 316 |
Orphan drugs |
|
21 CFR Part 50 |
Protection of human subjects |
|
21 CFR Part 56 |
IRBs |
|
21 CFR Part 201 |
Drug labelling |
|
21 CFR Part 54 |
Financial disclosure by clinical investigators |
Table 1: Federal set of laws to be valid towards of this IND Application method
The fixed reason of an IND is “to guarantee that subjects will not feature unwarranted danger of damage” in a clinical investigation that engages usage of the drug. As a result, towards approving drug evaluation in humans; this FDA requires enough information to measure the safety of the planned research study (Molzon, 2006). The IND is the method by which the investigator or sponsor delivers the mandatory information to get approval to manage an investigational representative toward human beings otherwise accepted medicine used for innovative sign otherwise on fresh inhabitants of patients. All evaluations with the purpose of usage of a drug are not conventionally designed for marketing with this FDA will always need of IND. With quiet lane set about classification intended for “innovative medicine” every single evaluation by not just innovative molecular entity otherwise not accepted pharmaceuticals other than accepted medicines using for not accepted suggestion, within novel formulations, during novel dosages, within long-suffering people to it will subsist on enlarged threat involve in IND. Under detailed criteria, an exception beginning from the IND necessity could be present to meet (Jacobs & Seifried, 2007).
Investigational new drug application
There are different IND. For individual sponsor-investigators, the IND will be considered as a “research IND”. Another group will be “commercial IND. FDA sort out IND application the same as “commercial” if a sponsor is a commercial object otherwise individual about this organization about this National organization about Health otherwise it was apparent that this medicine might be ultimately commercialized (Manning et al., 2020). FDA issues various regulations about filing IND. 80% of the Guidance’s deal with industry i.e., commercial.
Within the assured categories there is another designation. An “investigator IND” is a research IND agreed by an investigator who starts and performs the study with the instant control of this apply about the research medicine. This would characterize evaluations performed under the supervision of sponsor-investigators. Additional IND types include an “emergency IND” that lets the FDA permit for an untried drug in disaster situations that do not permit moment meant for file an IND or for patients who do not have access to the drug under a set of rules. Similarly, the “treatment IND” allows entry for subjects in serious circumstances to trial drugs that have revealed ability in early clinical testing but before final FDA review. Last, an “exploratory IND” is led early in phase 1types of research about an agent. These studies engage inadequate human being coverage and are designed without therapeutic intent and are initial to conducting more expressive traditional safety and lenience studies and allow for greater flexibility within this drug improvement procedure (Caldron et al., 2012).
Additionally, for antimicrobials, the FDA has a conference program to facilitate interactions among the sponsor and the FDA before filing an IND involving the action of bacterial, fungal, and viral infections, opportunistic infections, emerging infections (including naturally emerging diseases and potential bio threat agents), topical microbicide directed at avoidance about HIV spread, and shift refusal.
General principles
The common system for an IND embraces providing evidence animal pharmacology and toxicology studies, manufacturing data, and clinical protocols and investigator information. The goal is to offer FDA details on the way to agree on evaluation to assure safety about the contestant. To support sponsor-investigators naturally, this IND determination does not cause similar wide-ranging particulars together with preclinical evaluations otherwise manufacturing and development information as would be essential for a profitable sponsor pertains to IND in favor of until now unapproved medicine, particularly near the beginning of development (Hynes & Buckwalter, 2016). Toward the particular period, this will be because these studies performed by sponsor-investigators frequently use FDA-accepted pharmaceuticals. The reminder is that sponsor-investigator has responsibilities while together sponsor and investigator, and investigations performed below designation are recurrently single-site studies.
Investigational new drug guidance and planning
To support marketable sponsors, this medicine development is faraway extra multifaceted and implicated method contrast through sponsor-investigator. Analogously, the pre-IND method will be extra noble and frequently require programmed meetings otherwise teleconference. In favor of sponsor-investigator, the majority question is classically not as complex. Though, being investigators supposed to create and make use of this organization property. FDA net has forms that can be downloaded, descriptions about IND survey method, and schedule about rules resting on finale about outline and secretarial needs. FDA issues rules with the intention of IND giving in method especially sponsor-investigators (Freireich, 2006). A wide-ranging particular for sponsors is to guide preclinical and phase 1 evaluations and pre-IND session be furthermore planned. FDA builds contact details in favor of CDER and CBER official obtainable going on FDA net. Quires of IND system can head for suitable division, usually with telephone otherwise email.
FDA form 1571
IND demand, FDA Form 1571, proposes an association lying on the technique near this information with the planned research. By definition, the sponsor is the single individual starting and captivating task for the study. For this reason, the individual investigators who begin and perform trials assemble this criterion about a sponsor-investigator. Reminder especially, if any pharmaceutical group will provide medicines otherwise placebos, this individual investigator will be still named as a sponsor. To support sponsor-investigators, an element about the particulars essential lying on 1571 extend beyond and was enclosed with this FDA Form 1572.
Form 1571 was utilized through every one of the applicants, profitable otherwise research investigators, and here there are many sections that are not applicable towards sponsor-investigator. In the same way, as this IND beginning sponsor-investigator engage application about FDA-approved medicine, many replies within this 1571 were strong, changed, otherwise still mislaid assess through pharmaceutical industry sponsor (Buchneva, 2018). Entirely, the designation was not required in favor of Phase about Research (section 8), IND Number (section 6) was remind empty through primary purpose, Contract Research Organization (section 13) be supposed to be marked as “no,” and contact information for sponsor representative (sections 18 and 19) is reminding empty. As a result, this series numeral was “0000” by the original request (section 10). Ensuing IND alteration enlarges sequential amount through 1 within the order about assent.
Because research medicine was a commercially obtainable product, the information necessary through FDA was customized for a sponsor-investigator evaluation through the industrial sponsor. But marketed medicine is utilized “with no alteration toward permitted wrapping,” this 1571 must include brand name, generic name, quantity type, power, and the group number. Drug Master Files (21 CFR 314.42), Product License Applications (21 CFR Part 601), otherwise Investigator’s Brochure (IB) will be not needed (Swanson, 2015).
If manufactured goods are supplied within a non-approved form, afterward manufacturing and controls particulars, pharmacology and toxicology data, otherwise, details beginning preceding human research is needed, if not to details have earlier than been proposed toward FDA. But this is the case, and then a means for the FDA to reference the preceding information will be needed. Typically, this is done through note since the novel sponsor approves the right of market entry and includes case recognition number. But this dosage form is customized by the investigator, after that developing and the particulates of the controls, pharmacology and toxicology information, otherwise, information beginning previous human researches might be needed. Conversation along with FDA will assist clarity by which extra data was required.
Section permitted. “Contents about Application”, is likewise shortened for most sponsor-investigator proposals. For this section, items 2, 3, and 4 (Table of Contents, Introductory declaration, and General Investigational Plan, respectively) may be addressed in the cover letter. The evidence detail is not dissimilar from evidence obligatory by most local IRBs. Projects by commercially obtainable pharmaceutical goods to be utilized exclusive of modification can effectively express through orientation toward normal identifiers. “Environmental Assessment” can address through an unconditional elimination report. Though, if this pharmaceutical mediator was customized by any means, extra particulars are needed. Every part of developing particulars must succumb (Gawai et al., 2017). Similarly, as representatives have this possible meant for medicine addiction otherwise mistreatment, if it is radioactive, otherwise condition is that is utilized during pediatric evaluations, extra particulars might be required. The extra complete account about particulars needed existed within 21 CFR 312.23.
For every part of the form within the IND application, the sponsor-investigator was an individual human being answerable used for this behavior, development, assessment, and estimation about wellbeing related through trial. The significant one is to make total and consistent contact information for all forms and correspondence. Correspondence attends to and telephone number scheduled be supposed to as well specify the majority successful contact particulars meant for individual sponsor-investigator, together with a morning telephone number.
Here is a small dissimilarity within finishing and proposing 1571 meant for CBER than for CDER. Due to the extremely dedicated character about CBER evaluations, investigators are supposed to check with CBER openly to support regulation (Nahler, 2009).
FDA form 1572
This form comprises the “Statement of Investigator.” The particulars demanded about investigator’s qualifications and contact particulars can frequently exist meet by academic course vitae, noting lying on this form with the intention of particular was enclosed within the attachment. Sponsor-investigator rules about FDA Form 1572 persuade particulars needs meant for detailed sections of FDA Form 1571 (Novack, 2005).
FDA form 3674
IND application should exist to depart with documentation that the necessities about section 402(j) of the Public Health Service Act will be satisfied. The United States Public Law 110-85 (FDA Amendments Act of 2007), Title VIII, Section 801, needs registration of “applicable clinical trials.” Every restricted scientific investigation to utilize medicine regulated by FDA should have to be registered with the omission of phase-1 studies. The intention of the legislation requiring that appropriate clinical studies be registered was on the way to create confidence that the public has a right to use information about convinced clinical trials that are being conducted, including access and results. This listing procedure was performed through filing test particulars along with Protocol Registration organization about clinical trials scurry through US National Library of Medicine by National Institutes of Health. Form 3674 certification needs proper ClinicalTrials.gov identifiers to be attained as of registration. FDA has concerned outline supervision resting on this certification system (Moore et al., 2014).
On behalf of a sponsor-investigator filing IND, this accountability is meant for register rest through the investigator. For the most of sponsor-investigators, an institution through regulatory mistake intended for-perform about evaluation been possibly previously registered to investigate thing through registration account, and investigator no need toward make a detach registration relation.
Submitting an IND
The cover memo must depart through IND submission. Comprise detection about sponsor-investigator, a clear suggestion that this is an initial IND submission and ensure to facilitate contact particulars is clear and complete. Because this is the initial IND submission, there is no IND number. Each chronological association concerning IND must obtain chronological recognizing sequential digit, by which primary capitulation, will be as “0000.” Confirm label about evaluation. Pay attention towards these contact particulars accurately equal within 1571 and 1572 toward avoiding some postponement within communications. Because this method is time-sensitive, postponement because of communications faults can have some major penalties. Fling submission toward awareness about partition supervises therapeutic region designed for the medicine study. But they have a conversation among an individual at CDER otherwise CBER, will express submission to a definite receiver. The IND must be surrendered in 3 copies, in which, 1 is novel and 2 copies. Special binders and packaging are not needed (Jha et al., 2021). Submission addresses for the IND application is given in table 2.
|
IND submissions to CDER: For a Drug: |
Food and Drug Administration Center for Drug Evaluation and Research Central Document Room 5901-B Ammendale Rd Beltsville, MD 20705-1266 |
|
IND submissions to CDER: For a therapeutic biological product |
Food and Drug Administration Center for Drug Evaluation and Research Therapeutic Biological Products Document Room 5901-B Ammendale Rd Beltsville, MD 20705-1266 |
|
IND submissions to CBER: For a Biological Product: |
Center for Biologics Evaluation and Research HFM-99, Room 200N |
Table 2: IND application submission addresses
Following receipt of IND by the FDA
The IND will be routed to the appropriate division for review. A memo about acceptance is forwarded to the sponsor-investigator. This memo gives consign IND number, date established, and name and telephone number about FDA project manager toward whom quires relating to the application and additional communication must be guided. This IND will become effectual after 30 days from the affirmed FDA acknowledgment date except for FDA fling warning if not done. FDA normally will not file a memo informing the sponsor-investigator about endorsement. Evaluations will start after the 30 days, but FDA will not advise the investigator otherwise. But FDA desires additional particulars otherwise elucidating a 30-day window will not be pretentious except FDA provides a suggestion that the research was conducted completely otherwise incomplete clinical grasp. Limited grasp makes agree to a definite fraction about research toward starting whereas additional element will not get started. A scientific grasp clearly explains intending to research could not get started.
Reaction towards clinical hold
A clinical hold occurs when the FDA associates the sponsor-investigator and shows that the study cannot start pending decisions of questions. These exact queries organization has to be transmitting on the way to the investigator, normally via telephone followed via comprehensive memo. Leading acceptance about the listing of FDA distress, sponsor-investigator must respond toward problems mentioned within the memo within the total. This cover memo will go together with the responsibility must point out the reply through the title, Clinical Hold Complete Response. Similarly, this supplementary FDA Form 1571 must point out through sequential digit and checkbox as will be answer headed for clinical hold. The clock lying on the evaluation method will not start in anticipation of every issue that has been addressed and replies had been established and recognized through FDA.
FDA should respond in 30 days from acceptance about the total reply from the sponsor-investigator. The agency will concern a letter that lifts the clinical hold, areas of the study on partial hold, or that the study continues to be on hold pending resolution of ongoing questions. Until the FDA shows that a hold has been detached and research should not continue (Degnan et al., 2012).
Regulatory requirements for an IND during study and at completion
After compliance with IND the regulatory process under which a study progresses. Here are some enduring obligations with the aim of sponsor-investigator who have the same opinion toward the autograph of FDA Form 1571. Briefly, sponsor-investigator concurs on the way to maintain IND in progress, on the way to alert FDA regarding protection problem, toward folder yearly information, and on the way to inform FDA as soon as research complete meant for some cause. Several amendments toward IND must be filed along with FDA (Di 2001).
Protocol Amendments (21 CFR 312.30)
Requirements in this sector permitted intended for filing a novel procedure, modification toward the procedure, otherwise adding a fresh sleuth. Vicissitudes can include any upsurge or decline in drug contact by dose or period, a change in the subject populace’s addition or barring, or a change in nursing for defense. The IRB with misunderstanding dependability must similarly be notified and give sanction. The amended protocols must have succumbed before execution with the exception of a protocol change intended to remove an apparent immediate hazard to subjects. Here in this folder, IRB was informing here according to a set of laws and the FDA consequently advised (Chiodin et al., 2019).
Information Amendments (21 CFR 312.31)
Related to change within this investigation procedure, adjustments within the essential information concerning IND to facilitate which are not in extra evidence will be added through information modification. This can comprise changes in toxicology, chemistry, otherwise other technical information. All adjustments must be obviously tagged while toward contents (E.g., Information Amendment: Pharmacology- Toxicology). Adjustments are supposed to not be concerned extra commonly than that of the 30-days (Holbein, 2009).
Safety Reports (21 CFR 312.32)
Sponsor-investigators are answerable for investigating all protection apprehension brings toward notice. They should report FDA, every part of joining investigators, and restricted IRB about adverse knowledge associated by making use of medicine with the intention of mutually grave and unpredicted within written IND wellbeing information. These uniformly apply toward several judgments to suggest important hazards in favor of human being subjects. The time boundary designed for submitting was not more than that of 15 calendar days following the sponsor’s primary acceptance of details. The report is supposed to finish through FDA Form 3500A or else within the narrative set-up. This description must have tagged “IND Safety Report.” Sponsor-investigator was answerable in favor of evaluating the importance of description within the perspective of further wellbeing information.
If the case is any bereavement otherwise serious knowledge interconnected through research medicine, warning about FDA should exist not over 7 days from the sponsor-investigator’s primary unloading about particulars. This must be completed either through telephone otherwise through a true copy program (Jarow et al., 2016).
Annual Reports (21 CFR 312.33)
This should be filed by the sponsor investigator. Maximum Filing time is bounded within 60 days from the yearly time of IND. If it contains a variety of sets of rules in distinct IND, each one must be there recognized through title and have a summary report. The investigator should include grades about every research still within the development and every research concluded throughout earlier years. Progress of enrollment should be tallied plus the total number of subjects considered, the number entered to date, quantity of subjects whose contribution in the study was finished as designed; and the number who fallen exposed to research meant for several causes. If research had finished, or else if short-term consequences identified, a tense report of results should be integrated. A review of every IND protection reports surrendered throughout the precedent year is supposed to be integrated. Summary about several major adjustments within the pharmacology, toxicology, or else technical information must be incorporated. Finally, the map on behalf of the upcoming year must be confirmed (Browne, 1997).
Observing responsibilities for sponsor-investigators
Checking about research is an ongoing responsibility. The regulations honestly charge the sponsor-investigator with responsibility. Sponsors must supervise and guarantee that human subjects are sufficiently protected, that all reported clinical data are precise and complete and that this performing about the trial is in agreement with the protocol and regulations. Unique to drug studies is the added dependability for drug responsibility. Investigators should moreover proper every trouble to happen throughout research or else finish research besides informing to IRB, FDA, and additional investigators (Lee, 2005).
Conclusions
Gathering all the regulatory necessities meant for performing medicine research was an important part of conducting clinical research. Filing in addition to preserve IND might appear as intimidating. Although sponsor-investigator, functioning among FDA, can convene the regulatory requirements in addition to that are able to proceed with their research study with minimal delay. FDA composes it simple on the way to get in touch with the colonels who are accountable for managing of the IND. The supervision for filing the essential documents is inclusive and willingly accessible from the FDA net.

Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org