International Journal of Medical Case Reports and Medical Research
OPEN ACCESS | Volume 5 - Issue 1 - 2026
ISSN No: 2994-6905 | Journal DOI: 10.61148/2994-6905/IJMCRMR


Mohammed Alhamood1*, Tareq Muhammad2, Mohammad Alshara3, Yaqoob Merhej4, Muhammad Dayoub5, Amin Abbass6 and Yasser Bader7
1Department of Neurology, Tishreen Military, Hospital, Damascus, Syria.
2Department of Rheumatology, Tishreen Military Hospital, Damascus, Syria.
3Department of Cardiology, Tishreen Military Hospital, Damascus, Syria.
4Department of Cardiology, Tishreen Military Hospital, Damascus, Syria.
5Laboratory Department, Tishreen Military Hospital, Damascus, Syria.
6Head of the Neurology Department at Military Medical, Services Administration, Tishreen Military Hospital, Damascus, Syrian Arab, Republic.
7Department of Neurology, Tishreen Military, Hospital, Damascus, Syria.
*Corresponding Author: Mohammed Alhamood, Department of Neurology, Tishreen Military, Hospital, Damascus, Syria.
Received Date: August 22, 2024
Accepted Date: September 04, 2024
Published Date: September 10, 2024
Citation: Mohammed Alhamood, Tareq Muhammad, Mohammad Alshara, Yaqoob Merhej and Muhammad Dayoub.et.al. (2024) “Case Report: Unveiling the Hidden Culprit: The First Documented Case of Fabry Disease in Syria Revealed Through Atypical Cardiac Symptoms.”, International Journal of Medical Case Reports and Medical Research, 3(4); DOI: 10.61148/2994-6905/IJMCRMR/057.
Copyright: © 2024. Mohammed Alhamood. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Fabry (FD) disease is an X-linked genetic disorder that results in the intracellular accumulation of glycosphingolipids, leading to renal, cardiac, and cerebrovascular complications. This case report underscores the importance of considering FD in patients presenting with atypical cardiac manifestations, such as asymmetric septal hypertrophy. Early diagnosis and intervention are crucial in managing FD and preventing further complications, particularly those affecting the heart and kidneys. Our patient's case highlights the need for increased awareness and screening for FD, especially in regions where the disease may be underdiagnosed due to limited resources. Despite the unavailability of enzyme replacement therapy, the patient's cardiac outcomes improved with the prescribed treatment regimen, although renal disease progression was noted. This first documented case of FD in Syria emphasizes the necessity for healthcare systems worldwide to recognize and address the complexities of this rare genetic disorder. Continued monitoring and support for patients with FD are essential to improve their quality of life and long-term prognosis.
Introduction:
Fabry disease (FD) is an X-linked genetic disorder characterized by a deficiency in the enzyme alpha-galactosidase A (α-GLA), leading to the intracellular accumulation of glycosphingolipids and subsequent renal, cardiac, and cerebrovascular complications [1]. The incidence rate is approximately 1 in 40,000 live births [2]. Cardiac symptoms are observed in about 60% of males and 50% of females with FD [3]. Left ventricular hypertrophy (LVH) is a common manifestation of Fabry cardiomyopathy, affecting up to 43% of males and 26% of females. LVH due to Fabry disease is typically concentric and symmetrical, with asymmetric septal hypertrophy being a very rare occurrence [4,5]. Approximately 0.9% of hypertrophic cardiomyopathy (HCM) cases are attributed to Fabry disease [6]. We present a very rare case of Fabry disease manifesting as asymmetric cardiac septal hypertrophy, which is also the first documented case of Fabry disease in Syria.
Diagnosing Fabry disease can be a significant challenge in resource-limited settings due to a lack of awareness and limited access to advanced diagnostic tests. Early diagnosis and intervention are crucial in managing the disease and preventing further complications, particularly those affecting the heart and kidneys. Increasing awareness and implementing early screening programs can contribute to improving patient outcomes in these regions.
Case Presentation:
A 33-year-old Middle East man arrived at the emergency department complaining of chest discomfort and palpitations that began 15 minutes prior. Two months earlier, he had an unexplained episode of atrial fibrillation that resolved on its own. Since childhood, he had experienced burning and tingling sensations in his hands and feet, along with joint and muscle pain after physical activity. These symptoms had been variously attributed to growing pains and fibromyalgia by several physicians. During adolescence, he also suffered from unexplained acroparesthesia, reduced sweating (hypohidrosis), and intolerance to heat. His family history revealed that his 36-year-old brother had experienced similar burning pain in the hands and feet since childhood, along with episodes of abdominal pain, diarrhea, and paroxysmal atrial fibrillation. Additionally, his 62-year-old uncle and two cousins had suffered from chronic kidney failure starting in their third decade of life. Upon physical examination, his blood pressure was 140/90 mmHg, heart rate 70 bpm, respiratory rate 18 breaths per minute, and body temperature 36.5°C.
A 12-lead electrocardiogram (ECG) showed atrial fibrillation with a controlled ventricular response, which spontaneously resolved within 30 minutes. The patient was subsequently admitted to the cardiology department for further evaluation.
Transthoracic echocardiography revealed asymmetric septal hypertrophy measuring 16.3 mm. The left ventricular ejection fraction was recorded at 65% (see Figure 1). Laboratory analysis indicated a 24-hour urine protein level of 1300 mg/day (reference range: 0-150 mg/day), serum creatinine at 1.09 mg/dL (normal range: 0.5 – 1.3 mg/dL), and creatinine clearance at 76 mL/min (normal range for males: 97 to 137 mL/min). Troponin-I levels were measured at 0.1 ng/mL (normal range: 0 – 0.04 ng/mL).
Other laboratory results fell within normal parameters. The chest X-ray appeared normal. Both the 24-hour blood pressure monitoring and Holter monitor readings were unremarkable.
An abdominal ultrasound identified multiple renal cysts, with the largest measuring up to 2 cm (see Figure 2). Ophthalmic examination revealed angiokeratomas on the right eyelid, conjunctival vascular tortuosity, and an aneurysm (see Figure 3).

Figure 1: Transthoracic echocardiography revealed asymmetric septal hypertrophy up to 16.3 mm.
1-A: parasternal long axis view M Mode showing asymmetric septal.
hypertrophy in basal septum.
1-B: parasternal long axis view 2D showing asymmetric septal hypertrophy.
in basal septum.
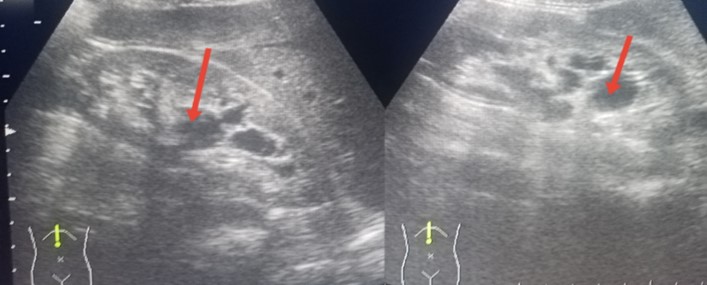
Figure 2: An abdominal ultrasound showing multiple renal cysts in both kidneys.
2-A: The right kidney with cysts. 2-B: The left kidney with cysts.

Figure 3: Ophthalmic examination
3-A: conjunctival vascular tortuosity. 3-B: Angiokeratomas on the right eyelid.
Given the patient's presentation of left LVH, proteinuria, angiokeratoma on the eyelid, acroparesthesia, hypohidrosis, and episodic joint pain, combined with his significant family medical history, we suspected FD. To confirm the diagnosis, α-GLA levels were measured, revealing a deficiency with levels at 0.6 μM/h (reference range: >3.31 μM/h). Consequently, the patient was diagnosed with FD and commenced on a treatment regimen including perindopril 2 mg/day, bisoprolol fumarate 2.5 mg, and apixaban 5 mg. Six months later, upon re-evaluation, there were no new episodes of atrial fibrillation, and transthoracic echocardiography indicated no progression of the cardiac disease. However, laboratory tests revealed an increase in 24-hour urine protein to 2170 mg/day. The patient was advised to continue biannual monitoring.
Discussion:
FD presents with a variety of clinical symptoms, typically beginning in childhood, though diagnosis is often delayed, leading to damage in vital organs. Newborn screenings have revealed that the actual prevalence of FD is higher than previously estimated [7]. Most research on FD originates from high-income countries such as the United States, several European nations, Japan, and South Korea, where financial resources are a key driver of research activity. It is crucial for health systems worldwide to be aware of FD and its complications. Early diagnosis is essential to prevent complications, particularly those affecting the kidneys and heart [8].
Cardiac involvement is the leading cause of death in Fabry patients [2], with manifestations including ventricular hypertrophy, valve thickening, heart failure, angina, cardiac conduction abnormalities, and sudden death. Hypertrophic cardiomyopathy (HCM) is the most common cardiac manifestation of FD [5], and 0.9% of HCM cases are secondary to FD, necessitating its exclusion in all HCM patients [6]. LVH due to FD is common but rarely presents as asymmetric septal hypertrophy [4-5].
Echocardiography is the primary investigation for suspecting and managing FD, offering an effective, non-invasive, rapid, and accessible method for assessing cardiac involvement [9]. In our case, transthoracic echocardiography revealed asymmetrical septal hypertrophy, and the presence of hypohidrosis, acroparesthesia, angiokeratoma, and proteinuria, along with a notable family history, heightened the suspicion of FD. Lysosomal testing confirmed a deficiency of alpha-galactosidase A, sufficient for diagnosing FD in males [10]. Following recommendations, our patient was treated with perindopril, bisoprolol fumarate, and apixaban [11, 12], though enzyme replacement therapy (ERT) was unavailable. A six-month re-evaluation showed improved cardiac outcomes but progression of renal disease. A-GLA test on the patient's brother revealed a result of 0.4 μM/h, leading to a diagnosis of FD, though financial constraints prevented testing other family members. This case is the first reported instance of FD in Syria, notable for its atypical cardiac manifestation.
Genetic Analysis:
Due to resource limitations in our country, it was not feasible to perform the genetic analysis to identify the α-GLA gene mutation. Additionally, conducting this analysis abroad was not an option due to the prohibitive costs involved. The absence of this genetic information poses a challenge in fully understanding the case and enhancing the scientific value of the manuscript.،
Clinical Implications:
Early diagnosis of FD can significantly improve patient outcomes by preventing severe complications. This case underscores the necessity for heightened clinical suspicion in patients with unexplained left ventricular hypertrophy. Implementing screening guidelines for FD in similar cases could lead to earlier detection and better management, ultimately improving patient prognosis.
Treatment Challenges and Alternative Strategies:
In the absence of ERT, patients face significant challenges in managing the symptoms and complications associated with Fabry disease. In this case report, an alternative treatment strategy was chosen, including the use of medications such as perindopril, bisoprolol fumarate, and apixaban to manage cardiac symptoms and prevent complications. Further research is necessary to identify alternative treatment options that are effective and affordable in resource-limited settings.
Conclusion:
This case report highlights the importance of considering Fabry disease (FD) in patients presenting with atypical cardiac manifestations, such as asymmetric septal hypertrophy. Early diagnosis and intervention are crucial in managing FD and preventing further complications, particularly those affecting the heart and kidneys. Our patient's case underscores the need for increased awareness and screening for FD, especially in regions where the disease may be underdiagnosed due to limited resources. Despite the unavailability of enzyme replacement therapy (ERT), the patient's cardiac outcomes improved with the prescribed treatment regimen, although renal disease progression was noted.
This first documented case of FD in Syria emphasizes the necessity for healthcare systems worldwide to recognize and address the complexities of this rare genetic disorder. Continued monitoring and support for patients with FD are essential to improve their quality of life and long-term prognosis.
Acknowledgements:
Our heartfelt thanks go out to the Military Medical Services Administration, Dr. Ammar Suleiman, and the administration of Tishreen Military Hospital, especially Dr. Moufid Darwich, that great man and comprehensive beacon who lights our path towards knowledge and science. He is the pride of our hospital and a medical symbol we cherish. We are deeply grateful to the compassionate and dedicated medical staff at the hospital, as well as the doctors from the Ophthalmology Department at Tishreen Military Hospital who provided essential data and expertise that led to completing this work.
Author Contributions:
Mohammed Alhamood: Reviewed the literature, wrote and revised the. manuscript, supervised the academic and scientific aspects of the manuscript, and performed grammar and language editing.
Tareq Muhammad: Reviewed the literature, wrote and revised the manuscript, Provided medical treatment and wrote the manuscript.
Mohammad Alshara: Provided medical treatment and wrote the manuscript.
Yaqoob Merhej: Provided medical treatment and wrote the manuscript.
Muhammad Dayoub: Provided medical treatment and wrote the manuscript.
Amin Abbass: Provided medical treatment and wrote the manuscript.
Yasser Bader: Provided medical treatment and wrote the manuscript.
Conflict of Interests:
There were no conflicts of interest.
Funding:
There was no source of funding for this study.
Ethical Approval:
No ethical approval was needed.
Consent:
Written informed consent was obtained from the patient for publication and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org