Clinical Research and Clinical Case Reports
OPEN ACCESS | Volume 7 - Issue 1 - 2026
ISSN No: 2836-2667 | Journal DOI: 10.61148/2836-2667/CRCCR


Hèla Ben Jmaà1, Mohamed Seddik1, Faten Dhouib1, Fatma Mhiri1, Hanene Abida2, Aiman Dammak1, Imed Frikha1.
1Department of cardiovascular and thoracic surgery Habib Bourguiba Hospital Sfax Tunisia. Faculty of medicine university of Sfax Tunisia.
2Department of anesthesiology Habib Bourguiba Hospital Sfax Tunisia. Faculty of medicine university of Sfax Tunisia.
*Corresponding authors: Hèla B Jmaà, Department of cardiovascular and thoracic surgery Habib Bourguiba Hospital Sfax Tunisia.
Received Date: January 15, 2024
Accepted Date: January 19, 2024
Published Date: January 24, 2024
Citation: Hèla B Jmaà, Seddik M, Dhouib F, Mhiri F, Abida H, Dammak A, Frikha I, (2024). “Surgical management of familial aortic disease”. Clinical Research and Clinical Case Reports, 5(1); DOI: http;//doi.org/08.2024/1.1072.
Copyright: © 2024 Hèla B Jmaà. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Pregnancy may increase the risk of aortic dissection in young women genetically predisposed in the third trimester and the peri-partum period. This disease must be managed with emergency. Later, aortic familial diseases must be screened in the children by genetic tests, because of the high risk of rapid developing dissection or aneurysm in the childhood, whose management is challenging. We report the cases of a pregnant woman operated for aortic dissection, and her son operated four years later for aneurysm of the ascending aorta, and aortic insufficiency. Children with familial aortic root aneurysm may have intervention of valve sparing and aortic replacement if the aortic regurgitation is minimal. But, long-term clinical and echocardiographic follow-up is mandatory.
Introduction:
In patients with familial aortopathy, aortic enlargement during pregnancy is more pronounced, leading to a more rapid increase in the overall aortic diameter with an increased risk of aortic dissection in the third trimester of pregnancy, which is a frequent cause of maternal death [1, 2]. Hereditary aortic disease includes a clinically and genetically heterogeneous group of disorders [3]. These familial aneurysms progressively enlarge and predispose to acute aortic dissections in all the family members. So, regular follow-up and screening of these disorders in the children is mandatory, in order to indicate surgery in the optimal moment.We report two clinical cases of aortic hereditary disease managed surgically with success.
Case report 1:
A 38-year-old woman in her thirty-sixth week of pregnancy, who had one ten-year old male child, was admitted to our department for sudden chest pain and dyspnea.Her medical history was unremarkable, except for a family history of a sudden death in one brother, but no diagnosed connective tissue disease. Her blood pressure was 160/85 mm Hg. Electrocardiogram revealed normal sinus rhythm without repolarization anomalies. D-Dimers dosage was normal.
Computed tomography scan was conducted and diagnosed Stanford type A aortic dissection, and the diameter of the ascending aorta was 47 mm (figure 1).
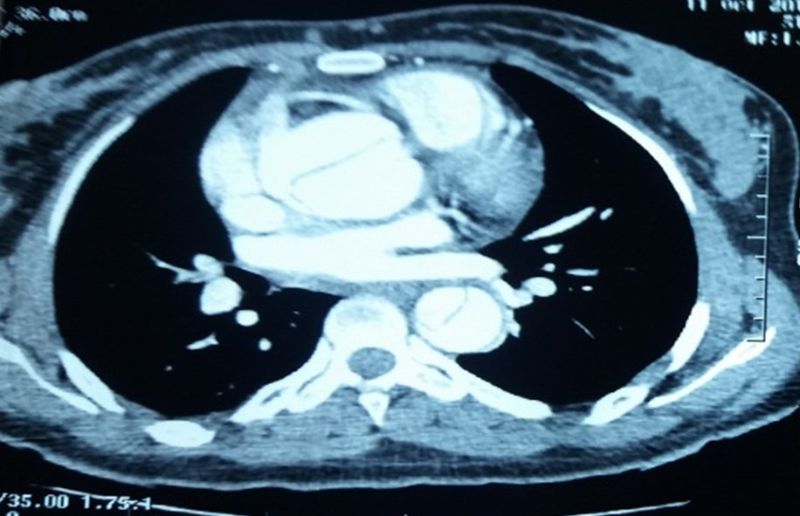
Figure 1: CT scan showing a dissection of the thoracic aorta.
The patient underwent urgent surgery. The name of the operation was cesarean and Bentall procedure. A live female infant was delivered. After removal of the aortic clamp, the heart automatically resumed, and the cardiopulmonary bypass was successfully terminated after the left ventricular assist (figures 2 and 3).
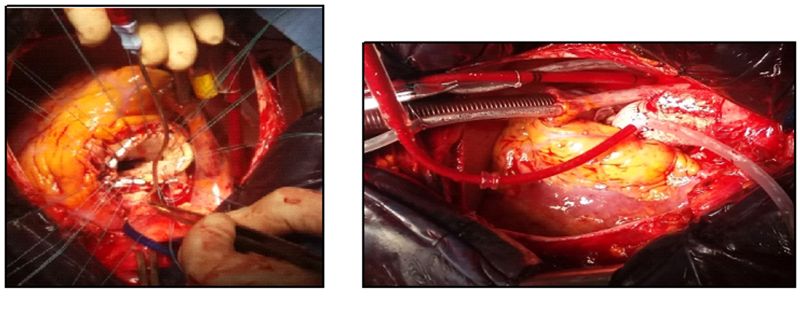 Figures 2 and 3: Intraoperative views revealing the replacement of the aortic valve and the ascending aorta.
Figures 2 and 3: Intraoperative views revealing the replacement of the aortic valve and the ascending aorta.
The tracheal intubation was removed 24 hours after surgery, and she was followed up after the operation, and both the patient and the infant survived.The family members of the patient were addressed for genetic consultation, but genes identification cannot be performed in our country. So, they had clinical, echocardiographic and scanographic follow-up in the pediatric department.
Case report 2:
Four years later, her fourteen-year-old son, with 185 cm of height and 80 kg of weight, was asymptomatic. He had regular screening of aortic disease by echocardiography.
His chest radiography showed scoliosis (figure 4).
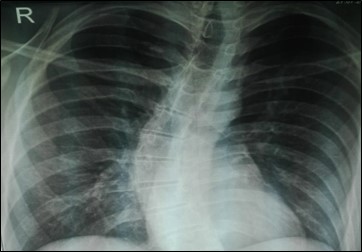
Figure 4: Chest radiography of the second patient showing scoliosis.
Echocardiography revealed a dilation of the ascending aorta: the diameter of the sinuses of Valsalva was 50 mm, the diameter of the sino-tubular junction was 52 mm, and the diameter of the segment 1 was 37 mm (figure 5).
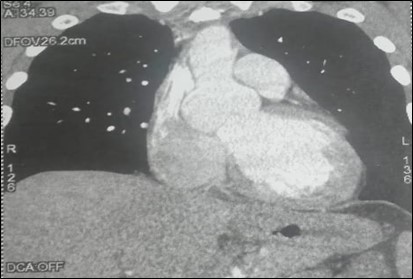
Figure 5: CT scan showing a dilation of the ascending aorta of the second patient.
The patient underwent conservative surgery of the aortic valve with replacement of the ascending aorta, and reimplantation of the coronary arteries (Tyrone David technique) (figures 6 and 7).
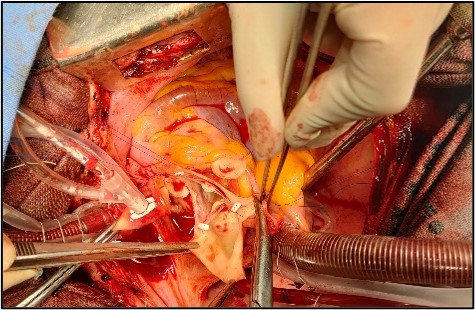

Figures 6 and 7: Intraoperative views revealing the aortic annuloplasty with replacement of the ascending aorta.
The postoperative course of the patient was uneventful.
Discussion:
The association of pregnancy with increased risk of aortic dissection may be caused by the hormonal changes, the increased circulating volume and cardiac output, and pre-existing blood hypertension leading to hemodynamic stress on the aortic wall, particularly those in the peri-partum period [4].Also, the risk of aortic complications associated with pregnancy in women with Marfan syndrome and other heritable connective tissue disorders, is related to the lack of knowledge of underlying diagnosis and potential presence of large aortic aneurysm prior to pregnancy, such as the case of our first patient whose aortic root diameter was 47 mm [5, 1].In a retrospective study of members of the Dutch Association of Marfan patients, 5 of 44 had a peri-partum aortic dissection. In total, 2 of the 4 Marfan syndrome women who had a type A dissection, were unaware of their diagnosis prior to the index event [6]. For women at 28 weeks or more gestational age, and suffer from type A aortic dissection, cesarean section followed by urgent repair of the dissection might be more appropriate [7]. That was the strategy we adopted for our first patient. whereas non-surgical management is preferred for acute type B aortic dissection [8]. Heritable aortic disease is defined as thoracic aortic enlargement and aortic wall weakness, with evidence of progression to the life-threatening complication of acute aortic dissection or rupture with fatal consequences. The altered gene triggering disease can be used to identify family members at risk [3]. There are syndromic and non-syndromic presentations. FBN1 is well established as the causal gene in patients with Marfan syndrome [9]. The FBN2 gene, underlying congenital contractural arachnodactyly, has also been isolated in children with mild, and non-progressive aortic dilatation [10]. There has been less research on Loeys–Dietz syndrome. Compared with the non-pregnant population, most aortic dissections during pregnancy are genetically triggered [11]. The thoracic aortic disease had to be clinically significant in terms of progressive aortic enlargement and/or dissection to trigger routine aortic imaging, medical and surgical management to prevent aortic dissections or other vascular complications, and predictive genetic testing of at-risk family members [3]. This genetic information might prevent future aortic events in the probands and family members who are at risk of acute aortic dissection [12]. Approximately 15% of patients with aortic aneurysm have a first-degree relative with an aortic aneurysm, and segregation analyses of such families have suggested a major gene defect [13]. This aneurysm screening in the family members of our first patient, who has a familial history of sudden death, has permitted to diagnose the aortic root aneurysm in his son at the age of fourteen years (second patient). Genes identification cannot be performed in our country.These aortic root aneurysms are rare in children. They are usually associated with connective tissue diseases or congenital heart anomalies. In children with Marfan syndrome, Fraser and colleagues [14] proceed with surgical intervention for aortic root aneurysms once the annular diameter has reached 5 cm or if the aneurysm is enlarging more than 0.5 cm per year.Aortic root replacement may be indicated in these children to prevent risk of aneurysm rupture, and aortic dissection. Guidelines for techniques and timing of surgery have been advocated, according to the size of the aortic root and the severity of the connective tissue disorder. The Bentall procedure, which was the gold standard, carries two potential drawbacks: need for long-term anticoagulation and difficulty in implanting an adult-size composite conduit in small children [15]. Valve-sparing root replacement has evolved through the pioneering work of surgeons such as David and colleagues [16]. The benefits of conserving the valve are avoidance of complications associated with anticoagulation therapy and prostheses. However, reoperation may be necessary for progressive aortic regurgitation, due to sub-commissural triangle stretching and absence of annular stabilization. So, strict follow-up is mandatory for these children. Fraser and colleagues [14], present single-institution data from a 20-year period describing 100 consecutive cases of valve-sparing aortic root replacement in patients aged less than 18 years. The majority of the patients had underlying connective tissue disease, such as Marfan syndrome (51%) or Loeys–Dietz syndrome (39%). Urbanski [17] considered that adaptive root repair in isolated sinus replacement is an effective and long-lasting method of valve-sparing repair in selected patients with marfanoid habitus.
Conclusion:
Pregnancy in Marfan syndrome and other hereditary aortic diseases women is associated with an increased risk of dissection in the peri-partum period. Current advances in the genetics of these diseases led to an early screening of aortic aneurysms and dissection in their family members, especially their children. The management of large aortic root aneurysm in children is challenging. Valve-sparing root replacement is a safe and effective option for these patients, like the case of our second patient. Because of late aortic insufficiency and pseudo-aneurysm formation, long follow-up on aortopathy in pediatric patients with connective tissue disorder will undoubtedly aid in defining surgical timing.
Conflict of interest: no conflict of interest












Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org