Clinical Medical Case Reports and Case Series
OPEN ACCESS | Volume 2 - Issue 1 - 2025
ISSN No: 3065-7644 | Journal DOI: 10.61148/3065-7644/CMCRCS


Elkhoyaali Adil
Elkhoyaali.a, military training hospital mohamed v rabat, morocco
*Corresponding author: Elkhoyaali Adil, Elkhoyaali.a, military training hospital mohamed v rabat, morocco.
Received: May 03, 2025
Accepted: May 30, 2025
Published: June 03, 2025
Citation: Elkhoyaali Adil. (2025) “X-Linked Retinitis Pigmentosa Revealed by Tapetal Like Reflex: When You All Need for Diagnosis Is a Fundus Picture- A Case Report and Littérature Review.”, Clinical Medical Case Reports and Case Series, 2(1); DOI: 10.61148/3065-7644/CMCRCS/012
Copyright: © 2025. Elkhoyaali Adil. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Tapetal-like reflex (TLR) is an unusual, golden, bright, scintillating, particulate reflection on indirect ophthalmoscopy that relatively spares the fovea, similar to that seen in the eyes of some vertebrates.1 TLR has been described in female carriers of X-linked RP and has also been seen in a healthy young male.2
Introduction:
Retinitis pigmentosa is a group of inherited retinal diseases characterized by degeneration of rod and cone photoreceptors3.Human inherited retinal degenerations are genetically heterogeneous, with well over 100 genes implicated to date.4 Measurements of retinal function, such as the electroretinogram, show that photoreceptor function is usually compromised many years before symptomatic night blindness, visual field scotomas, or decreased visual acuity occur. More than 45 genes have been identified for retinitis pigmentosa.5,6
X-linked Retinitis Pigmentosa GTPase Regulator is a GTPase-binding protein encoded in humans by the RPGR gene, which is located on the X chromosome and is commonly associated with X-linked retinitis pigmentosa (XLRP)7. In photoreceptor cells, RPGR is localized in the connecting cilium, which connects the protein-synthesizing inner segment to the photosensitive outer segment and is involved in modulating cargo trafficking between the two segments.8
Case Presentation:
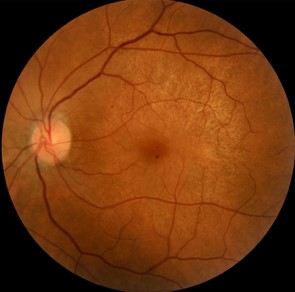
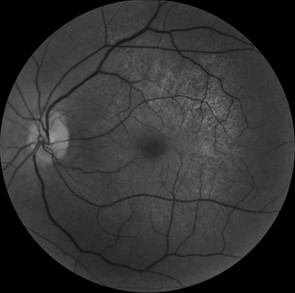
We report the case of a 45-year-old woman presented with complaints of decreased vision in the left eye (LE). Her best-corrected visual acuity in the RE was 10/10 and left eye was 06/10. Both eyes (BEs) anterior segment examination was unremarkable. There was no history of night blindness or decreased vision in any of the family members. Fundus examination revealed presence of retinal pigment epithelial (RPE) hypopigmentation and atrophy at the posterior pole along with an enhanced golden tapetal sheen seen in posterior pole of the left eye (figure 1A). Fundus autofluorescence revealed a crescent-shaped hyperautofluorescence in LE (figure 1B). Spectral domain optical coherence tomography (SD-OCT) revealed a normal foveal contour in the left eye with thining of photoreceptor layer to the macula in LE with small drusens(figure 2). Based on the multimodal imaging findings, we suspected a starting manifestations of retinitis pigmentosa and the patient was advised for genetic analysis revealing mutation on x-linked RPGR/RP3 GENE then set for observation and a routine follow-up.
A B
Figure1: A: fundus photo showing tapetal like reflex in left eye B: fundus autofluorescence showing hyperreflectivity of the posterior pole by atrophy starting of the retinal pigmented epithelium
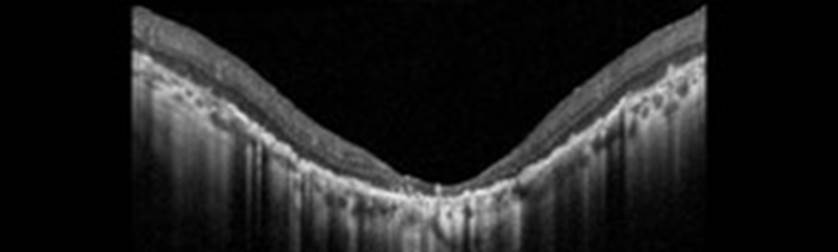
Figure 2: macular oct of left eye showing extreme thining of photoreceptor layer with small drusens
Discussion:
TLR has been described in female carriers of X-linked RP and has also been observed in a healthy young male.3 In addition, abnormal fundus reflections in male patients have been reported in Oguchi disease, X-linked retinoschisis, cone-shaped retinal dystrophy, and early X-linked RP. The TLR has been reported to be located deep to the retinal vasculature and at the level of the outer retina and RPE.3 It may be due to deposits, thickening or degeneration of Bruch's membrane, retinal deposits or an alteration at the level of the RPE-photoreceptor interface.3 We report the presence of an enhanced TLR located in the midperiphery in a patient with sector RP, confirming its association with inherited retinal degenerations; however, the nature and origin of the phenomenon are still not clear and further studies will shed light on this unique finding.
The worldwide prevalence of RP is 1:3000 to 1:7000 including simple, syndromic and systemic disease. There is usually no gender predilection, but males are slightly more affected than females due to the existence of X-linked RP as seen in our patient. RP has no ethnic specificity, but in certain forms related to specific gene mutations it is more common in conanginous populations (such as the USH3 gene associated with Usher syndrome type III).9
Most cases of retinitis pigmentosa are monogenic, but the disease is still very genetically heterogeneous10. Researchers have identified at least 45 loci at which mutations cause the disease, and these genes together account for the disease in just over half of all patients.11
Since the first report describing linkage of an RP locus to a DNA marker on human chromosome X in 1984 12, more than 40 genes have been associated with RP. Non-syndromic or "simple" cases may be inherited as autosomal dominant (20-25%), autosomal recessive (15-20%), X-linked recessive (10-15%), or sporadic/simple (30%), and 5% may be early-onset and grouped as part of Leber congenital amaurosis (LCA). Rarer forms also exist, such as X-linked dominant, mitochondrial, and digenic (due to mutations in two different genes).9
XLRP is due to mutation of 6 genes located in chromosome X, but only two have been identified so far: the retinitis pigmentosa gtpase regulator RPGR and the Rp-2 protein RP2.13The mutations in these 2 genes are reposabale for 80/100 of clinical cases of XLRP which make them a good target to small molecules drug and gene therapy approaches.14
Conclusion:
Tapetal like reflex is frequently an incidental fundiscopic finding associated with many retinal diseases like XLRP and may appear years before symptoms and vision loss.
Well understanding of the phenotypes and genes included in RP is the key to be more efficient in developing new treatments of the disease including gene specific approaches,transplantation replacing retinal loss tissue and implanting electrical devices.


Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org