Clinical Medical Case Reports and Case Series
OPEN ACCESS | Volume 1 - Issue 2 - 2026
ISSN No: 3065-7644 | Journal DOI: 10.61148/3065-7644/CMCRCS


Priyanka V Kashyap1*, Dharmendra Singh2, Mayur K Bhat2, Naman Sahu2
1Department of Neurology AIIMS Bhopal.
2Senior Resident, Neurology.
*Corresponding author: Priyanka V Kashyap, Associate Professor & Incharge, Department of Neurology AIIMS Bhopal.
Received: June 15, 2026 | Accepted: June 22, 2026 | Published: June 26, 2026
Citation: Priyanka V Kashyap, Singh D, Mayur K Bhat, Sahu N. (2026) “Clinico-Etiological & Imaging spectrum of Hypertrophic Pachymeningitis – A single centre case series Report”, Clinical Medical Case Reports and Case Series, 3(2); DOI: 10.61148/3065-7644/CMCRCS/064.
Copyright: © 2026. Priyanka V Kashyap. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background: Hypertrophic pachymeningitis (HP) is a rare disorder of the central nervous system marked by inflammation and thickening of the dura mater, which may be localized or widespread. In many cases, the process can also extend to involve the leptomeninges and structures such as the tentorium. Patients mostly present with headache as the initial symptom, while other manifestations include cranial nerve palsies, seizures, and hearing loss.
Purpose: To study the clinical presentation, underlying etiologies, and radiological characteristics of patients with hypertrophic pachymeningitis managed at a tertiary care center at AIIMS Bhopal.
Methods: This descriptive case series consists of six patients diagnosed with hypertrophic pachymeningitis, based on characteristic neuroimaging features and supported by cerebrospinal fluid (CSF) and histopathological findings. Demographic characteristics, clinical presentation, examination findings, laboratory and radiological investigations, treatment, and short-term outcomes were collected from clinical records.
Results: Six patients (four females and two males, aged 18–50 years) were identified. Two patients had tubercular HP, another two had IgG4-related HP (IgG4-RHP), and remaining two were classified as idiopathic HP (IHP). Headache and cranial nerve involvement were the most frequent clinical features. Tubercular HP cases showed lymphocytic pleocytosis with raised protein and low CSF glucose, and CSF Cartridge Based Nucleic Acid Amplification Test (CBNAAT) positivity. In IgG4- RHP, one patient had markedly elevated serum IgG4 levels and both showed supportive histopathology. Magnetic Resonance Imaging (MRI) typically demonstrated diffuse or focal pachymeningeal thickening with contrast enhancement, often involving the cavernous sinus, tentorium, and posterior fossa. Tubercular HP patients responded well to antitubercular therapy with corticosteroids, IgG4-RHP cases improved with steroids and rituximab, and IHP patients responded to corticosteroids with azathioprine as a steroid sparing agent.
Conclusion: HP is an uncommon rarely suspected cause of chronic headache and cranial nerve palsies. A comprehensive etiological evaluation, including CSF analysis, serum IgG4 levels, detailed MRI, and histopathology whenever feasible, is essential to differentiate various forms as their management and prognosis differ. Early, cause-specific treatment can lead to favorable clinical outcomes.
Hypertrophic pachymeningitis (HP); IgG4-RHP; Idiopathic HP; Tubercular HP; Rituximab therapy
Hypertrophic pachymeningitis (HP) is a rare disorder of the central nervous system marked by inflammation and thickening of the dura mater, which may be localized or widespread. It may involve leptomeninges and tentorium as well. Incidence is approximately 0.949 per 100,000 individuals (1,2). Etiopathologically, HP can be either idiopathic or secondary to chronic infections such as tuberculosis, malignancies such as lymphoma, autoimmune disorders such as rheumatoid arthritis, and other systemic diseases including IgG4-related disease. Idiopathic hypertrophic pachymeningitis is considered only when all alternative etiologies have been rigorously excluded. (3–5). Pathophysiologically, Clinical manifestations results from venous congestion, ischemia from cortical vessel compression, and direct inflammatory infiltration of the brain parenchyma. Headache serves as the most common presenting symptom. Cranial nerve palsies is the second most frequent manifestation followed by seizures, hearing loss, and movement disorders. Diagnosis primarily relies on neuroimaging findings, supplemented by histopathological confirmation (6–8). IgG4-related hypertrophic pachymeningitis (IgG4-RHP) is a major secondary cause of this inflammatory disease (IgG4-RD) (5,9,10), in which the immune system drives a buildup of IgG4-positive plasma cells in affected tissues. Over time, this causes characteristic scarring in a whorl-like pattern called storiform fibrosis, and inflammation around veins causing obliterative phlebitis. (5,9,10). Serum IgG4 is often elevated in 70%–90% of IgG4-related patients, but it can be normal or only mildly increased in isolated IgG4-related hypertrophic pachymeningitis (IgG4-RHP). Meningeal biopsy remains the gold standard for diagnosing IgG4-related hypertrophic pachymeningitis (IgG4-RHP) (9,10). Management of HP is directed by the underlying cause. There are currently no consensus guidelines for the treatment of IgG-RHP and Idopathic hypertrophic pachymeningitis (IHP), although corticosteroids and other immunosuppressants have been used successfully in previous case reports and series (4,6–8). Some patients may require surgical decompression. Rituximab, a B-cell–depleting therapy, is a useful steroid-sparing option in IgG4-related disease and has been shown to improve symptoms, reduce serum IgG4 levels, and control many systemic manifestations. (9,10). The authors intend to present this case series of six patients suffering from hypertrophic pachymeningitis (HP), emphasising the uniqueness of slow sequential neural axis affection and rarely suspected if not aware of this entity.
Methods: This is a descriptive case series of six patients diagnosed with HP at a tertiary care neurology center. Patients having clinical features suggestive of HP and MRI evidence of focal or diffuse pachymeningeal thickening with contrast enhancement, with or without leptomeningeal involvement were included. Demographic details, presenting symptoms, neurological examination findings, laboratory investigations (including inflammatory markers, autoimmune profile, serum ACE, serum IgG4, and relevant infectious work‑up), CSF analysis, neuroimaging findings, histopathology, etiological diagnosis, treatment protocols, and short-term clinical outcomes were retrieved from hospital records. Etiological classification into tubercular HP, IgG4-RHP, and IHP was based on clinical features, imaging, CSF and microbiological data (including CBNAAT), serology (including serum IgG4), systemic evaluation, and, where available, meningeal biopsy.
Case summaries
Case 1: Tubercular hypertrophic pachymeningitis presenting with headache and proptosis
A 24-year-old woman presented with migrainous headache for 5 years that lasted for 10–12 hours. Over the last 2.5 years, it transformed into a continuous dull aching left hemicranial headache persisting throughout the day, associated with nausea, vomiting, and transient visual obscurations (TVOs) and aggravated by bending forward. She also developed gradually worsening swelling of the left eye over the past 2.5 years. Later proptosis developed associated with pain on eye movements, watering, a gritty sensation, double vision, and near-complete loss of vision. Examination revealed complete ophthalmoplegia of the affected eye with fundus revealed Grade II disc edema bilaterally. Rest of the neurological examination including motor, sensory, and cerebellar was normal. Routine investigations showed (Table 1) raised ESR. Vitamin D3, vitamin B12, and folate levels were also low. MRI brain and orbit revealed asymmetric pachymeningeal thickening with multiple areas of venous sinus thrombosis, including left cavernous sinus thrombosis, suggestive of hypertrophic pachymeningitis, with old healed granulomatous lesions in the head of the right caudate nucleus and left medial frontal gyrus (Figure 1A). There was partially empty sella, crowding of the posterior fossa, and mild prominence of the perineural CSF space in the retro-ocular segment of both optic nerves. These findings were consistent with HP. Evaluation for secondary causes of HP was negative, including serum IgG4 levels, viral markers, Antinuclear Antibody (ANA), Antineutrophil Cytoplasmic Antibody (ANCA), 24-hour urinary calcium, and serum Angiotensin-Converting Enzyme (ACE). CSF showed lymphomononuclear pleocytosis, low glucose, and borderline high protein; routine fungal and mycobacterial studies were negative, while CSF CBNAAT and the tuberculin skin test were positive. Based on these findings, a diagnosis of tubercular hypertrophic pachymeningitis was made. She was managed with 3 days of intravenous pulse methylprednisolone (1 g/day), followed by oral steroids and antitubercular therapy, anticoagulation were added along with supplementation of vitamin B12 &vitamin D. She gradually improved at 1 month and 3 month follow up.
Case 2: IgG4 Related Hypertrophic Pachymeningitis responding to Rituximab therapy
A 50-year-old woman presented with bilateral progressive hearing loss of three years duartion along with headache, and bulbar palsy over next one year. Examination revealed bilateral facial, eighth, and ninth cranial nerve palsies suggesting polyneuritis cranialis. Her inflammatory markers (CRP) was elevated and serum IgG4 was markedly raised (65.30 g/dL). MRI showed diffuse sheet-like pachymeningeal thickening and enhancement in the posterior fossa extending to C2, with mass effect on posterior fossa structures, hydrocephalus (Figure1B). A diagnosis of IgG4-related hypertrophic pachymeningitis was made and meningeal biopsy demonstrated storiform fibrosis with lymphocytic infiltrates. She was started on intravenous steroids and rituximab. Two doses of rituximab 1 g were administered 14 days apart, after which her headache completely resolved. During the 6 months follow up, she has showed improvement in headache frequency and severity and marked resolution of craniopathy.
Case 3: Idiopathic hypertrophic pachymeningitis presenting with headache and seizure responding to steroids
A 27-year-old woman presented with acute onset frontal headache for 1 month. The headache was throbbing and moderate in intensity, lasting more than 3 hours with infrequent vomiting, photophobia, and phonophobia and some relief on sleeping. She also reported low grade fever for 1 month, with evening rise of temperature, 2 episodes of tonic clonic seizures 1 week apart. MRI brain performed was suggestive of hypertrophic pachymeningitis, with thick sheet like diffuse and asymmetrical leptomeningeal enhancement over the bilateral temporal convexities and tentorium cerebelli with compression of posterior fossa structures (Figure 2A). A previously ongoing inflammatory process was suspected. Ophthalmology evaluation was normal. Investigation revealed normal IgG4 level, non-reactive VDRL, ACE and negative autoimmune work- up. CSF analysis was also normal. She was managed with intravenous pulse methylprednisolone therapy for 5 days followed by Oral steroids and azathioprine were then started for long term immunosuppression. Meningeal biopsy could not be performed due to technical reasons. Based on MRI findings, negative etiological work-up, and response to steroids, a diagnosis of idiopathic hypertrophic pachymeningitis was concluded.
Case 4: Tubercular hypertrophic pachymeningitis treated with antitubercular therapy and steroids
A 45-year-old man presented with moderate intensity, migrainous type headache for 2 years duration and later developed right eye painful ophthalmoplegia featuring drooping of the right eyelid, pain and restricted eye movements associated with fever. Neurological examination showed right fourth and sixth cranial nerve palsies. There was 5th sensory impairement on right side over V1, V2 distribution. Other neuraxis examinations were normal. The patient was evaluated for infectious, inflammatory, and neoplastic causes. Investigations revealed elevated ESR and CRP. HRCT thorax showed fibro- atelectatic and fibro-calcific changes involving both upper lobes suggestive of tuberculosis. Mantoux test was strongly positive (22 mm at 48 hours). A guarded lumbar puncture and CSF analysis showed mild pleocytosis with lymphocytic predominance, elevated protein, and low CSF glucose relative to blood glucose. MRI brain done outside showed diffuse, irregular dural thickening and enhancement in basal cisterns and the suprasellar region, non-enhancing FLAIR hyperintense signal in the right temporal, parietal, and genu of corpus callosum regions, and encasement of the right optic nerve with involvement of the right orbital canal (Figure 2B). MRI brain with contrast and MR spectroscopy favored granulomatous etiology, with no choline peak. Total serum IgG4 level was normal. The combination of positive Mantoux test, HRCT thorax findings, CSF hypoglycorrhia and high protein with positive CBNAAT and clinical presentation favored tuberculosis. He was managed with anti-edema measures (intravenous mannitol, oral spironolactone) and supportive care. A final diagnosis of tubercular hypertrophic pachymeningitis was made, and antitubercular drugs added with dexamethasone steroid tapered over 8 weeks.
Case 5: Idiopathic hypertrophic pachymeningitis responding to azathioprine
A 50-year-old man presented with holocranial, dullaching headache for 3 months with constitutional features in form of loss of appetite, weight loss. The general physical and neurological examinations was normal. MRI brain with contrast revealed marked diffuse dural and leptomeningeal thickening and enhancement, with a small subependymal cystic lesion with mild surrounding edema and enhancement in the left anterior temporal lobe (Figure 3). Differential diagnoses of infectious, neoplastic, and autoimmune etiologies were considered. Routine investigation- Complete Blood counts (CBC) showed normocytic normochromic anemia suggesting chronic course. ESR was raised, Tumor markers were nnormal, ANA and ANCA, Rheumatoid factor were negative. CSF analysis including autoimmune panel, Serum IgG4 levels were within normal limits. CECT abdomen and thorax were normal. Patients underwent Meningeal biopsy from the left fronto-parietal region that showed thickened dura with focal areas of meningothelial cell proliferation adjacent to the thickened dura and focal aggregates of lymphomononuclear cells, consistent with chronic inflammatory pachymeningitis without a definitive secondary cause. A probable diagnosis of chronic granulomatous meningitis was kept and patient managed as Idiopathic HP with intravenous dexamethasone and symptoms responded and immunosuppressive agent- azathioprine was added.
Case 6: Young female with IgG4 related hypertrophic pachymeningitis treated with rituximab
A 19-year-old woman with encephalitis illness and seizures 3 years earlier presented with
chronic daily holocranial headache and one episode of seizure. She had TVOs also.
Examination revealed Cushingoid facies, right eye RAPD (relative afferent pupillary defect)
And bilateral disc edema. Rest of the neurological examination was normal. Her MRI Brain with contrast revealed meningeal thickening diffusely, lytic bone lesions in frontal bone and ethmoid sinus involvement (image not shown). ENT consultation was obtained; nasal swabs for malignant cells and fungal pathology were negative. Routine investigations including vasculitic and viral markers were normal. CSF revealed high pressures (24 cm H₂O) with all cytobiochemical parameters normal, CSF was negative for malignant cytology, fungal including cryptococcal antigen, CBNAAT. Sarcoid workup was also negative. Bone marrow aspiration and biopsy were also negative. Skeletal survey was normal. Serum protein electrophoresis, CT chest, and CT abdomen showed no evidence of disseminated infection, sarcoidosis, or malignancy. Serum IgG4 level was elevated (2.69 g/L). Meningeal biopsy showed brain parenchyma with scattered neurons in a dense fibrillary neuroglial matrix, with edema and meningeal congestion, without granulomas or malignancy. As her IgG4 levels were raised and she was partially responsive to steroids, so immunosuppressive therapy with intravenous rituximab was initiated to which she responded dramatically over next 6 months.
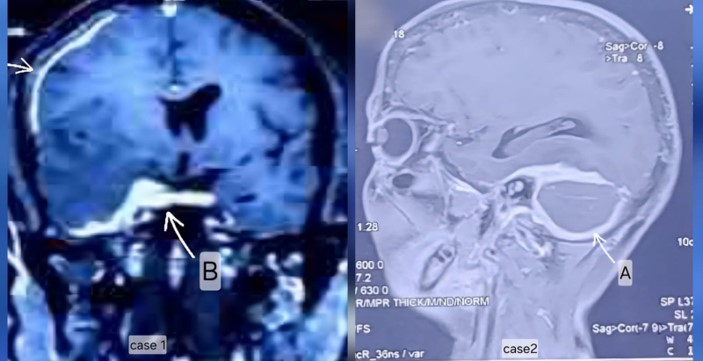
Figure 1A: (Case 1) MRI Brain T1 post contrast coronal section showing asymmetrical pachymeningeal thickening at multifocal area in (a)right frontoparietal and (b)temporal region
Figure 1B: (Case 2) MRI Brain T1 sagittal section with contrast Thick sheet like pachymeningeal thickening & enhancement in posterior fossa (A), extending into upper cervical spine region.
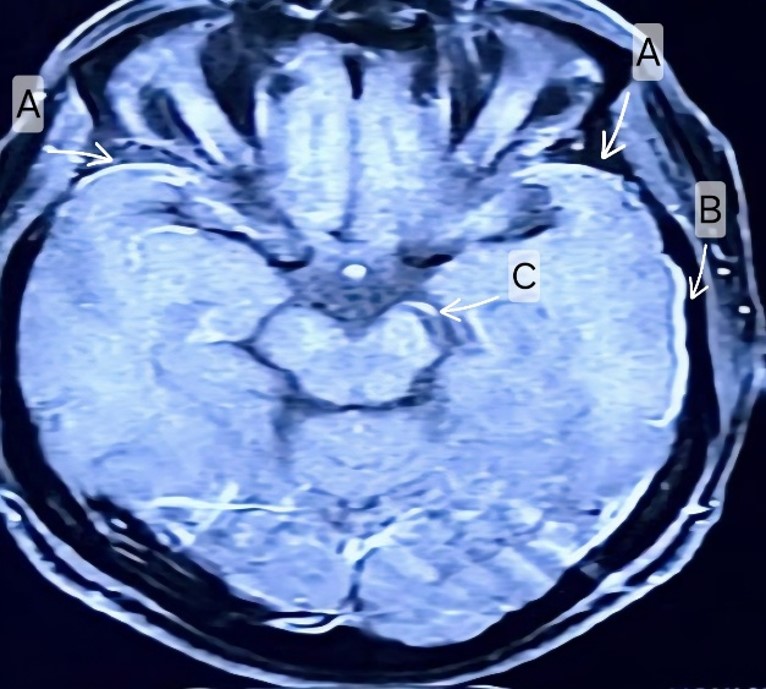
Figure 2A: Case 3: MRI Brain T1 contrast axial view showing Thick sheet like diffuse and asymmetrical leptomeningeal enhancement in bilateral temporal convexities (A, B), tentorium cerebelli (C)with contrast enhancement and compression over posterior fossa structures.
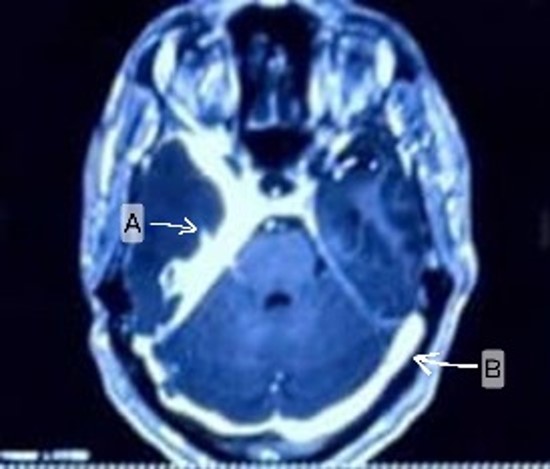
Figure 2B: Case 4 MRI Brain T1 contrast axial view showing (A, B) Diffuse, irregular thickening and dural enhancement in basal, suprasellar cisterns.
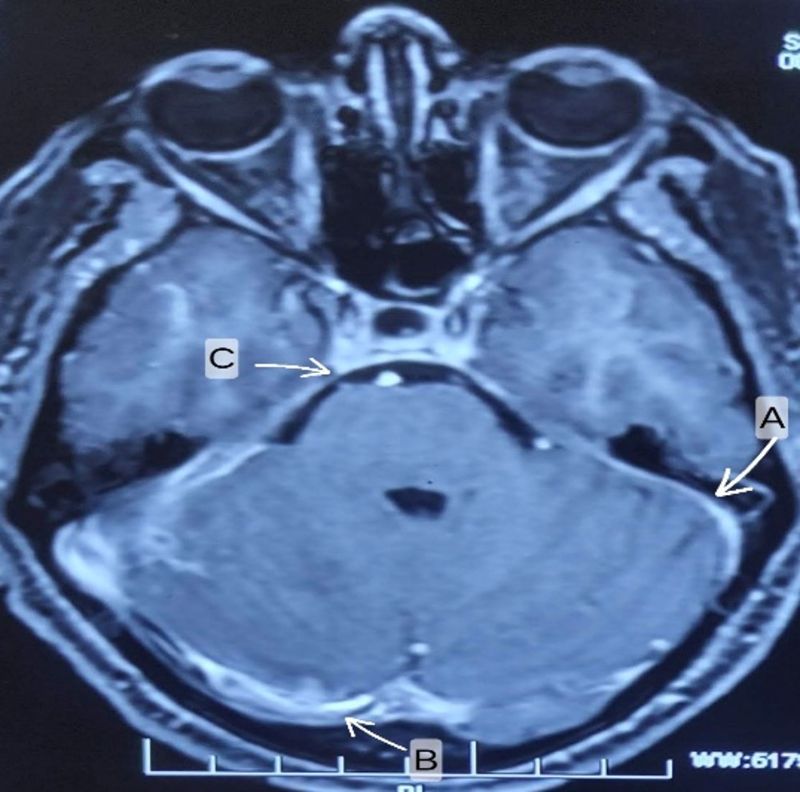
Figure 3: (Case 5)-MRI brain T1 contrast axial view (A, B, C arrows) shows Diffuse dural and leptomeningeal thickening with enhancement.
Table 1-Summary of Cases
|
Parameters |
Case 1 |
Case 2 |
Case 3 |
Case 4 |
Case 5 |
Case 6 |
|
CBC WBC HB Platelets |
10200 11.2 3,30,000 |
10730 9.2 3,32,000 |
9600 12 3,60,000 |
6400 13 3,20,000 |
11580 11.9 400000 |
7600 9 178000 |
|
LFT |
WNL |
WNL |
WNL |
WNL |
WNL |
WNL |
|
RFT |
WNL |
WNL |
WNL |
WNL |
WNL |
WNL |
|
CRP (mg/dl) |
3.52 |
43.01 |
1.2 |
63.4 |
61` |
142 |
|
ESR (mm/hour) |
37 |
51 |
43 |
80 |
42 |
37 |
|
ANA |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
|
ANCA |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
|
Chest-X-Ray |
WNL |
WNL |
WNL |
WNL |
WNL |
WNL |
|
S ACE Levels (U/L) |
18.3 |
14.6 |
20.1 |
18.4 |
17 |
9.5 |
|
IgG4 levels (g/L) |
0.57 |
65.30 |
0.88 |
2.7 |
0.65 |
2.6 |
|
24 Hours Urinary Calcium (mg/day) |
105 |
157 |
132 |
119 |
|
|
|
CSF study# Cells Protein Glucose Gram Stain AFB CBNAAT KOH India Ink
|
35 (L*) 93 41 Negative Negative Positive Negative Negative |
3(L*) 50 64 Negative Negative Negative Negative Negative |
47 76 Negative Negative Negative Negative Negative |
7(L*) 108 56 Negative Negative Positive Negative Negative |
4(L*) 49 62 Negative Negative Negative Negative Negative |
10(L*) 42 66 Negative Negative Negative Negative Negative |
|
HRCT Thorax |
Old fibrotic changes with Tree in Bud appearance in right upper lobe |
WNL |
WNL |
fibro atelectatic and fibrocalcific changes involving bilateral upper lobes, and few suspicious branching centrilobular nodules in right upper lobe suspicious of endobronchial infection. |
WNL |
WNL |
|
MRI Brain |
Asymmetrical pachymeningeal thickening with multiple areas of filling defects/ venous sinus thrombosis and left cavernous sinus thrombosis |
thick sheet like pachymeningeal thickening & enhancement in posterior fossa, extending into upper cervical spine region (till C2 vertebra) causing mass effect over posterior fossa structures, upstream hydrocephalus with compressive myelopathy at C1 level |
Thick sheet like diffuse and asymmetrical leptomeningeal enhancement in bilateral temporal convexities, tentorium cerebelli with contrast enhancement and compression over posterior fossa structures |
diffuse, irregular thickening and dural enhancement in basal, suprasellar cisterns, and non-enhancing FLAIR hyperintense signal in right temporal, parietal, GC region and encasement of right optic nerve and involvement of right orbital canal and fissure which explained the clinical deficits |
diffuse dural and leptomeningeal thickening and enhancement and a small subependymal cystic lesion with mild surrounding oedema and enhancement in left anterior temporal lobe brain parenchyma |
Pachymeningial thickening showing post contrast enhancement along bilateral frontal and temporal convexities, tentorium cerebelli, bilateral anterolateral cerebellar hemisphere rand posterior to clivus. |
|
Meningeal biopsy |
|
Storiform fibrosis with lymphocytic infiltrate |
|
|
|
Brain parenchyma with scattered neurons in dense and fibrillary neuroglial matrix with evidence of edema and meningeal congestion. |
*L- Lymphocytes, #CSF analysis reference values- Cells: 0-5 cells, Protein: 15-45 mg/dl, Glucose: 1/3rd to 2/3rd of Blood glucose level at time of Lumbar puncture, WNL-Within Normal Limits
CBC- complete blood counts, WBC- White blood counts, HB- Hemoglobin, LFT-Liver Function Tests, RFT- Renal function tests, CRP- C Reactive Protein, ANA- Antinuclear Antibody, ANCA- Antineutrophil cytoplasmic Antibody, ACE- Angiotensinogen Converting Enzyme.
Discussion
Hypertrophic pachymeningitis (HP) is a rare condition that needs high index of suspicion. This series is a collection of diagnosed proven cases of HP who present with very mundane clinical features that are missed in a busy neurology clinic if not kept in mind. Of the six cases, 4 were female and 2 were male, aged between 18 and 50 years. IHP and IgG4 RHP have been reported more in females as our series highlights (11). The etiological profile in our series revealed infective (2 cases- tubercular HP), granulomatous (2 were IgG4-RHP, and Idiopathic (2). Tuberculosis being endemic in India has a unique HP presentation with chronic phase without obvious eye-catching signs (12–14). Headache and cranial nerve involvement were the most common clinical phenotype in our series. Chronic, persistent headache of varying intensity is the most common initial symptom of HP associated with nausea and vomiting as reported by other clinicians (15–18). The mechanisms include inflammation of the duramater, mass effect on neural structures due to meningeal hypertrophy, venous sinus obstruction, and immune-mediated neuropathy. Cranial nerve involvement is the second most common symptom, with II and VII cranial nerves being predominantly affected (8,11,18). This is due to either nerve compression or the presence of an orbital pseudotumor, often involving optic nerve sometimes bilaterally (19-20). Some patients may present with fever and seizures as noted in our patients. (21–24). Anemia of chronic disease raised total leucocyte count, or elevated inflammatory markers may be present, as seen in one of our cases and in previous series. A detailed search for the infective /granulomatous on imaging of chest and abdomen (Chest X-ray, HRCT Cheat, CT abdomen) proves benchmark aid in haunting the diagnosis especially TB. The recommended investigative approach to such patients must include ANA, ANCA, rheumatoid factor, serum ACE level, and 24-hour urinary calcium level to rule out autoimmune disorders and sarcoidosis. CSF cytology, biochemistry, culture, CBNAAT, KOH mount, and India ink staining are helpful in diagnosing infectious causes. In this case series, tubercular cases showed lymphocytic pleocytosis with raised protein and low CSF glucose, and one case had CSF CBNAAT positivity, consistent with tubercular HP as also seen in previous reports (12–14). Approximately 70% of patients with HP exhibit intracranial hypertension with elevated leukocyte and protein levels in CSF. Aseptic lymphocytic meningitis may be seen in IgG4-RHP (21). Two of our cases had raised serum IgG4 levels and were diagnosed as IgG4-RHP. However, serum IgG4 is not a very sensitive marker, as it can be normal or only mildly elevated in some cases of IgG4-RD, and levels depend on the extent of organ involvement.
MR imagings findings of HP reveals thickening of the duramater that can be diffuse or focal, often involving the cavernous sinus and orbital apex. Contrast enhancement occurs as a consequence of ongoing inflammation and is reduced as the disease recovers. MRI plays a crucial role in distinguishing IHP from secondary HP. Secondary HP often exhibits dural thickening in the anterior and middle cranial fossae, whereas IHP typically shows homogeneous contrast enhancement (23,24). Additionally, hypointense signals on T2-weighted sequences and the T2 rim pattern (central T2 hyperintensity with a hypointense rim) strongly suggest HP. Treatment is based on the underlying cause. All infectious etiologies, particularly tuberculosis, should be vigorously ruled out before starting immunotherapy. Corticosteroids are the first line therapy in IgG4-RHP and IHP and shows robust response. If the patient respond partially or develop steroid dependence or side effects, clinician must consider immunosuppresive agents such as azathioprine, cyclosporine, or mycophenolate mofetil. Reports are favouring Rituximab as an off-label alternative. Our IgG4-RHP cases responded very well to rituximab, and idiopathic HP patient improved with azathioprine, observation alike other results (25,26). This series is unique representation of HP with vivid clinical phenotypes of headache and heterogeneous etiologies-infective, inflammatory and idiopathic and the role of steroids, immunsuppression in reversing the process along with disease specific drugs.
Conclusion
Hypertrophic pachymeningitis is a less suspected entity having very mundane presentations of chronic headpains, sometimes with or without cranial nerve affection. It must be kept as differentials in unresolving headaches, papilledema. MRI with contrast play Key role in diagnosing aided with CSF, immunological biomarkers tool including IgG4-RHP and IHP. If not diagnosed accurately can interfere badly with quality of life and add social burden. Early etiology specific treatment—antitubercular therapy for tubercular HP and immunosuppressive therapy (including rituximab) for IgG4-RHP and IHP can result in substantial clinical improvement and prevent irreversible neurological deficits.
Ethical considerations
Institutional Review Board approval is not required and conducted in accordance with the Declaration of Helsinki.
Consent: Written informed consent was obtained from the patients.
Acknowledgments
This case series is an original work and addresses an important clinical question in field/subspecialty and, we believe, offers findings of relevance to your readership. The manuscript is original, has not been published previously, and is not under consideration elsewhere. All authors have approved the final version and agree with its submission to your journal. We gratefully acknowledge the entire study team, the hospital staff, and, above all, the patients and their relatives, whose participation and trust made this research possible.
Authors contribution
PVK: Concept and design of the study, data acquisition, drafting of the manuscript.
DS: Data acquisition and analysis, interpretation of imaging, critical revision of the manuscript.
MKB: Writing, editing, references
NS: Clinical management of patients, data interpretation, manuscript review.
All authors read and approved the final manuscript.
Conflicts of Interest
No conflicts of interest to be declared by the authors
Consent to participate
Written informed consent was obtained from all individual participants (or their legal guardians) included in the study.
Consent for publication
Written informed consent for publication of anonymized clinical data and any accompanying images was obtained from all patients (or their legal guardians).
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not for profit sectors.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations:
ACE - ANGIOTENSIN-CONVERTING ENZYME
AFB - ACID-FAST BACILLI
ANA - ANTINUCLEAR ANTIBODY
ANCA - ANTINEUTROPHIL CYTOPLASMIC ANTIBODY
CBC - COMPLETE BLOOD COUNT
CBNAAT - CARTRIDGE BASED NUCLEIC ACID AMPLIFICATION TEST
CECT - CONTRAST-ENHANCED COMPUTED TOMOGRAPHY
CRP - C-REACTIVE PROTEIN
CSF - CEREBROSPINAL FLUID
ENT - EAR, NOSE, AND THROAT / OTORHINOLARYNGOLOGY
HB - HEMOGLOBIN
HBSAG - HEPATITIS B SURFACE ANTIGEN
HBV - HEPATITIS B VIRUS
HCV - HEPATITIS C VIRUS
HP - HYPERTROPHIC PACHYMENINGITIS
H₂O - WATER
IGG4‑RD - IGG4-RELATED DISEASE
IGG4‑RHP - IGG4-RELATED HYPERTROPHIC PACHYMENINGITIS
IHP - IDIOPATHIC HYPERTROPHIC PACHYMENINGITIS
KOH MOUNT - POTASSIUM HYDROXIDE MOUNT
LFT - LIVER FUNCTION TESTS
MMF - MYCOPHENOLATE MOFETIL
MRS - MAGNETIC RESONANCE SPECTROSCOPY
RFT - RENAL FUNCTION TESTS
RPR - RAPID PLASMA REAGIN
TVOS - TRANSIENT VISUAL OBSCURATION
WBC - WHITE BLOOD CELL COUNT
WNL - WITHIN NORMAL LIMIT


Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org