Clinical Case Reports and Clinical Study
OPEN ACCESS | Volume 12 - Issue 5 - 2025
ISSN No: 2766-8614 | Journal DOI: 10.61148/2766-8614/JCCRCS


July Mehta1, Shailesh Desai2*, Bhavna Vora3, Moumita Misra4
1Head operations- Metropolis Healthcare Limited, Mumbai, Maharashtra, India.
2Head operations- Metropolis healthcare limited, Mumbai, Maharashtra, India.
3Head operations- Metropolis healthcare limited, Mumbai, Maharashtra, India.
4Chief of Labs- Metropolis healthcare limited, Mumbai, Maharashtra, India.
*Corresponding author: Shailesh Desai, Head operations- Metropolis healthcare limited, Mumbai, Maharashtra, India.
Received: October 01, 2025 | Accepted: October 08, 2025 | Published: October 15, 2025
Citation: Mehta J, Desai S, Vora B, Misra M. (2025) “A Missed Diagnosis or Neglected Parental Premarital Counselling of Rare Case of Compound Heterozygous Sickle Cell- β+Thalassemia in Patient: Case Report” Clinical Case Reports and Clinical Study, 12(4); DOI: 10.61148/2766-8614/JCCRCS/217.
Copyright: © 2025 Shailesh Desai. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
The prevalence of abnormal haemoglobin genes is indeed a significant global health concern. Among these, compound heterozygous HbS β-thalassaemia is particularly rare, with a prevalence of 0.02% in India, and only a few single case reports have been documented.
Case report
We report a case of a 5- year-old male child was brought to us with history of fever and jaundice lasting one week with prior ICU hospitalization 6 months ago for similar symptoms. During his admission, symptomatic treatment was given, despite his elevated WBC count and elevated creatinine levels noted. We identified sickle cells, target cells, and NRBCs, along with low haemoglobin levels. Sickle cell anaemia diagnosed after 3 months of relapse, and with proper family history of parents, which was missed in initial hospitalization. It was concluded that he is having compound heterozygous HbS β-thalassaemia. Counselling would have been better option for his parents as his father have β-thalassemia trait and mother have HbS trait.
Conclusions
Distinguishing compound heterozygous haemoglobinopathies from isolated sickle cell anaemia or β-thalassaemia can be tricky due to their overlapping clinical and pathological features. However, accurate diagnosis is crucial for appropriate management and prognosis.
Compound heterozygous Sickle Cell- β+Thalassemia, Foetal haemoglobin (HbF), Haemoglobin A2 (HbA2), High performance liquid chromatography (HPLC).
Sickle cell disease is an inherited blood disorder caused by a mutation in the beta-globin gene, leading to the production of abnormal hemoglobin (HbS). This results in sickle-shaped red blood cells, which can cause blockages in blood vessels, leading to pain and complications. One variant, HbS/beta-thalassemia, occurs when the sickle cell gene is inherited alongside the beta-thalassemia gene. There are two types: Sickle cell β+ thalassemia wherein some beta-globin is produced and Sickle cell β0 thalassemia where no beta-globin is produced [1].
The severity of the disease depends on the amount of normal haemoglobin (HbA) present. Higher HbA levels result in milder symptoms, categorized into three types; Type-I: 1-7% HbA (more severe), Type-II: 7-14% HbA (moderate) and Type-III: 14-25% HbA (mild) [2]. Diagnosis involves clinical evaluation, blood tests, and high-performance liquid chromatography (HPLC) to differentiate between sickle cell variants and other hemoglobinopathies [3].
Case Report
A 5-year-old male child presented with abdominal pain, pallor, fever, and malena to the hospital ward. On clinical examination patient was afebrile, pulse of 109 per min, blood pressure of 90/60 mmHg with pallor (++). Systemic examination was normal with no hepatomegaly or splenomegaly being noted.
Laboratory testing showed low RBC Count (2.47 million/mm3; Normal: 4.5-6.0 million/mm3), low hemoglobin (Hb) (5.7 g/dl; Normal: 14-16 g/dl), low hematocrit (HCT) (17.8%; Normal: 42 ± 7%), low Platelet Count (0.10 lakhs/mm3; Normal: 1.5-4.5 lakhs/mm3), high White Blood Cell Count (53,400/mm3; Normal: 4,000-11,000/mm3). RBC morphology was abnormal with smear showing microcytosis, macrocytosis, hypochromia, anisocytosis, poikilocytosis, target cells, schistocytes [Figure 1]. Bleeding time and clotting time was normal.
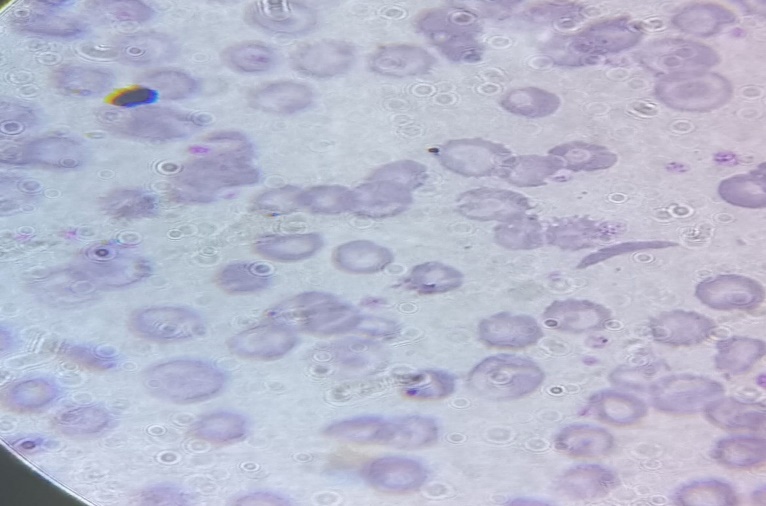
Figure 1: Peripheral smear shows Sickle Cell and Target Cells 100X.
Serum Bilirubin (Total): 5.30 mg/dl (Normal: 0.2 - 1.2 mg/dl), Serum Bilirubin (Direct): 2.30 mg/dl (Normal: 0.01 - 0.3 mg/dl), Serum Bilirubin (Indirect): 3.00 mg/dl (Normal: 0.2 - 0.8 mg/dl), Serum SGPT: 1450 Units/litre (Normal: 5 - 35 U/L) and Serum SGOT: 3070 Units/litre (Normal: 5 - 40 U/L).
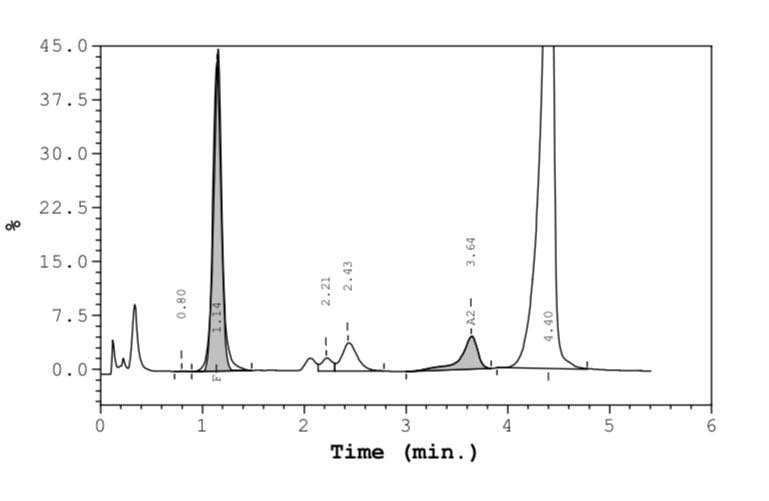
Figure 2: Chromatograms of male child showing compound heterozygous HbS β thalassaemia.
High-Performance Liquid Chromatography (HPLC) results showed Haemoglobin A2 (HbA2): 4.6% (suggestive of β-thalassaemia trait); Haemoglobin F (HbF): 21.2% (elevated, commonly seen in β-thalassaemia and sickle cell disease and Haemoglobin S (HbS): 70.0% (indicating sickling disorder) [Figure 2]. Sickling test was positive, confirming the presence of sickling abnormality. The results are highly suggestive of compound heterozygous sickle cell β-thalassaemia, requiring parental screening and molecular studies for definitive confirmation.
We did HPLC testing on Biorad analysers of D10 and also on Tosho G8 analysers. We could not find significant difference between the two analyses in terms of readings for each parameters.
Parental history was taken and no history of consangious marriage was given. Father is 37-year-old male and underwent laboratory testing. His hemoglobin (Hb): 12.4 g/dL was slightly lower, RBC Count: 6.46 million/mm³ was higher, PCV (Hematocrit): 40%, MCV & MCH was reduced suggestive of microcytic hypochromic anemia. HbA2 Level: 4.71% was above normal reference range of 2.0 - 3.5%. Hb HPLC analysis suggestive of Beta Thalassemia Trait [Figure 3].
Mother is a 28-year-old female and following laboratory parameters were observed: hemoglobin (Hb): 13.7 g/dL (Within normal range), RBC Count: 4.81 million/mm³ (At the upper limit of the normal range), PCV (Hematocrit): 41.3% (Normal), MCV & MCH: was within normal range. HbA Level: 39.99% which is lower than the normal reference range of 83.0 - 95.0%, HbA2 Level: 2.25% (Within normal range), Hb Sickle: 34.61% was significantly elevated, suggestive of HbS Trait [Figure 3].
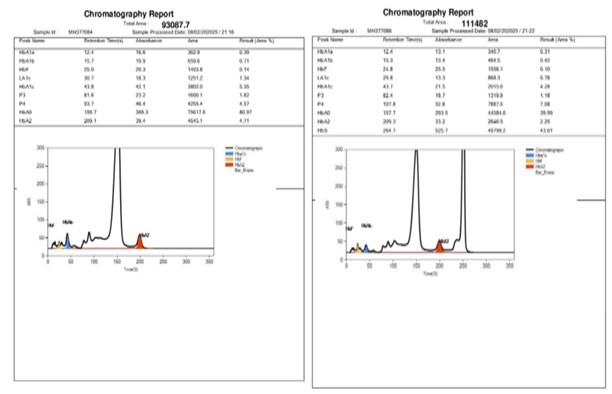
Figure 3: Chromatograms of father and mother showing with β thalassaemia trait and HbS trait, respectively.
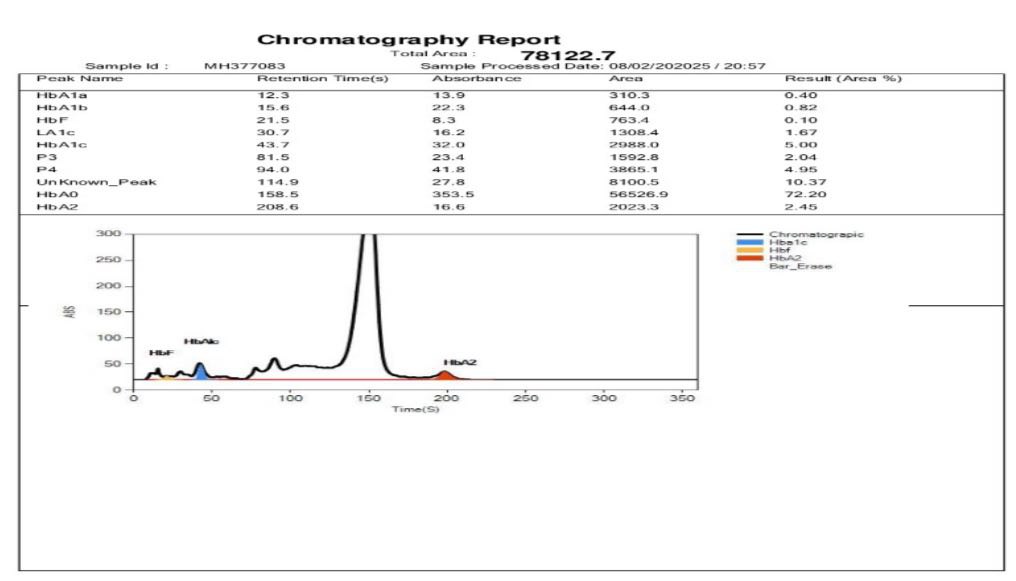
Figure 4: Chromatograms of female child showing normal hemoglobin with no anomaly detected.
Baby sister of the patient is 2 years old and she also underwent laboratory testing. Haemoglobin (Hb): 10.1 g/dL (Below normal reference range of 11.0-14.0 g/dL). RBC Count: 4.87 million/mm³ (Within normal range). PCV (Hematocrit): 32.6% (Slightly below
normal. MCV & MCH: Reduced (Suggestive of microcytic anemia). HbA Level: 72.20% (Below normal reference range of 83.0-95.0%), HbA2 Level: 2.45% (Within normal range) and HbF (Fetal Haemoglobin): 2.04% (Within normal range for children under 2 years). Hb HPLC Analysis: Normal Hb chromatographic pattern with no indication of Beta Thalassemia trait or common abnormal haemoglobin [Figure 4].
Molecular studies were advised but due to financial constraints it couldn’t be done. Patient was managed with hydroxyurea tablets along with folvite supplements. Patient is also advised to get bone marrow transplant which is still under consideration due to financial constraints. They were counselled for future precautions to prevent sickling crisis along with various genetic aspects related to the disease.
Discussion
According to WHO, approximately 5% of the world's population carries a potentially pathological haemoglobin gene. In India, the frequency of sickle cell disorders is around 4.3%, while β-thalassaemia occurs in 3-4% of the population. Among these, compound heterozygous HbS β-thalassaemia is particularly rare, with a prevalence of 0.02% in India, and only a few single case reports have been documented [1, 2, and 8]. This historical account of the HbS gene in India provides key insights into the geographical spread and genetic prevalence of sickle cell disease and related hemoglobinopathies.
The first documented case in an Irula boy from Nilgiri Hills marks the initial discovery, while its subsequent identification in the sickle cell belts of central India suggests a regional genetic clustering [11]. Additionally, the global presence of sickle cell-β thalassemia, including cases reported from Nepal, Kuwait, and India, highlights how hemoglobinopathies can vary based on population genetics. In our case, boy belonged to the North Eastern region in Maharashtra state of India which is rare as previously reported cases belonged to the North-eastern regions such as Assam, Nagaland, Manipur, Tripura and West Bengal. The study identifying 12 cases of double heterozygous HbS-β+ thalassemia further emphasizes the rarity and complexity of such genetic conditions [12].
Compound heterozygous HbS β-thalassemia presents a diagnostic challenge because its hematological features often overlap with thalassemia intermedia and homozygous sickle cell disease. The presence of target cells, basophilic stippling, microcytic hypochromic RBCs, and reduced MCV/MCH can closely resemble sickle cell anemia, making differentiation difficult using peripheral blood smears (PBS) alone as in our case [8]. HbA2 elevation is a key marker in identifying β-thalassemia carriers as seen in our case, but complications arise when HbF, HbA2, and HbA levels fall outside expected diagnostic thresholds [6]. This can make it challenging to distinguish between homozygous HbS (sickle cell anemia) and compound heterozygous HbS β-thalassemia based purely on laboratory results [4].
Agarwal, R. et al reported case which highlighted the diagnostic complexity of hemoglobinopathies, particularly when distinguishing between compound heterozygous HbS β-thalassemia and homozygous sickle cell disease. The severe anemia (Hb levels of 1.8 g/dL and 1.5 g/dL) in both children prompted further investigation via HPLC, which revealed significantly elevated HbS and HbF levels, raising suspicion of hemoglobinopathy. Since both differential diagnoses—HbS homozygous (sickle cell anemia) and compound heterozygous HbS β-thalassemia—were considered, a family study was conducted. The results confirmed the father carried β-thalassemia trait, while the mother carried sickle cell trait, leading to the final diagnosis of compound heterozygous HbS β-thalassemia in both children. This underscores the importance of family genetic evaluation in resolving hemoglobinopathy diagnostic dilemmas, as laboratory findings alone may sometimes be inconclusive [4].
In a study by Meliti A et al., demonstrated the importance of multidisciplinary collaboration in medicine—especially when clinical signs don't immediately suggest an underlying haematological condition. The presence of sickle cells in a fluid sample from the aneurysmal bone cyst was an unexpected finding in an eight-year-old boy presented to the hospital for evaluation of short stature, leading to immediate re-evaluation by specialists. This reinforces how hematological disorders, like sickle cell disease, can sometimes present atypically—causing complications beyond anemia, such as bone-related pathology. The swift coordination between orthopedics and haematology was crucial for refining the diagnosis. [5].
Priya J et al., emphasizes the diagnostic challenges in differentiating sickle cell anemia from compound heterozygous HbS β-thalassemia, particularly when peripheral blood smear (PBS) findings overlap. The presence of HbS >50% and a high HbF value (35.1%) initially led to two possible differential diagnoses. However, a family study confirmed that the patient inherited both sickle cell trait and β-thalassemia trait, making HbS β-thalassemia compound heterozygosity the final diagnosis. This case of 12-year-old boy highlights the importance of parental genetic evaluation, as distinguishing between these conditions is crucial for appropriate clinical management [6]. Our case showed the presence of HbS of 70%, HbF of 21.2% and HbA2 of 4.6% with initial diagnosis of sickle cell anemia and family history of thalassemia further confirmed the presence of compound heterozygous HbS β-thalassemia.
Chandra S et al. highlights the diagnostic complexity of hemoglobinopathies, particularly when sickle cell anemia and HbS β-thalassemia compound heterozygosity have overlapping hematological features. The patient is 16-year-old female presented with generalized weakness and intermittent jaundice were investigated through complete blood examination, revealing moderate microcytic hypochromic anemia along with anisocytosis and late normoblasts. Initially, HPLC analysis suggested sickle cell anemia, given the HbS >50% and normal HbA2 levels. However, a family study was conducted, which led to reclassification of the diagnosis—her brother was identified with sickle cell trait, while her sister was found to have β-thalassemia trait. This ultimately confirmed that the patient had compound heterozygous HbS β-thalassemia, rather than homozygous sickle cell disease [7].
Kaur M et al., reported case of 32-year-old male which underscores the importance of family studies in refining hematological diagnoses. While HbS >50% and normal HbA2 levels initially pointed toward sickle cell anemia, the parental and sibling evaluation helped clarify that the patient actually had compound heterozygous HbS β-thalassemia rather than homozygous sickle cell disease. This distinction is crucial, as the clinical manifestations of HbS β-thalassemia can differ significantly from classic sickle cell disease, potentially influencing treatment approaches and patient management [8].
Shrestha B et al., reported a case of five-year-old male who was initially diagnosed with sickle cell anemia based on an HbS level of 63.2%, but his healthy appearance and lack of transfusion history prompted further investigation. Hemoglobin electrophoresis (HPLC) can sometimes lead to reconsideration of a diagnosis, especially when clinical presentation doesn’t align with expected symptoms. The discovery that both parents had β-thalassemia trait and sickle cell trait suggests that the child may have compound heterozygosity rather than classic sickle cell disease [9].
Jain Mayank et al. reported case of 12-year-old boy of compound Sickle Beta Thalassemia with high foetal Haemoglobin; of Hereditary Persistence of Foetal Haemoglobin (HPFH). The high percentage of HbF makes it even a rarer case which has not been reported in literature so far [10].
This case studies and reports concludes that the critical role of family studies and molecular analysis in resolving diagnostic uncertainties in hemoglobinopathies. While peripheral blood smear (PBS) and HPLC findings can raise suspicion of rare cases like compound heterozygous HbS β-thalassemia, they may not always be definitive. When molecular studies are unavailable, family genetic evaluation serves as a reliable and accessible method for confirming diagnoses. This approach has been essential in cases where haematological features overlap, making differentiation between conditions like HbS β-thalassemia and sickle cell anemia challenging.
Conclusions
We hope to lead to more awareness among physicians that premarital counselling plays a crucial role in preventing the transmission of sickle cell disease and beta thalassemia to future generations. Since both conditions are hereditary, individuals who carry these genetic traits—heterozygotes—may unknowingly pass them on to their children, leading to severe health complications. This includes genetic awareness, risk assessment, reproductive options like prenatal diagnosis, preimplantation genetic testing (PGT), or opting for non-carrier partners and informed decision making by counselling.
Differentiating sickle cell-β+ thalassemia from sickle cell trait hinges on recognizing a higher proportion of HbS compared to HbA. This distinction is crucial in clinical diagnostics, as HbF and residual HbA tend to ameliorate disease severity in HbS-β+ thalassemia, unlike HbS-β⁰ thalassemia and homozygous sickle cell disease, which often present more severe complications.
Financial support and sponsorship
Metropolis healthcare limited supported financially to publish article.
Conflicts of Interest
The authors declare that they have no conflict of interests. Informed written consent for publication was obtained from the patient prior to collecting data and relevant medical history.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org