Aditum Journal of Clinical and Biomedical Research
OPEN ACCESS | Volume 8 - Issue 1 - 2026
ISSN No: 2993-9968 | Journal DOI: 10.61148/2993-9968/AJCBR


Ahmet Esat Göner, Hatice Esra Duran*
Department of Medical Biochemistry, Faculty of Medicine, Kafkas University, Kars, 36100, Turkey.
*Corresponding author: Assoc. Prof. Hatice Esra DURAN, Department of Basic Medical Sciences, Faculty of Medicine, Kafkas University, 36100/Kars, Turkey, Phone: +90 474 225 1150-7599. E-mail address: haticeesra4990@gmail.com, ORCID ID: 0000-0003-2080-0091.
Received: February 10, 2026 | Accepted: February 25, 2026 | Published: March 02, 2026
Citation: Ahmet E Göner, Hatice E Duran., (2026) “Strong Inhibition of Aldose Reductase: A Study on Pyrimidines” Aditum Journal of Clinical and Biomedical Research, 8(1); DOI: 10.61148/2993-9968/AJCBR/120.
Copyright: ©2026. Hatice Esra Duran. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Aldose reductase (ALR2; AKR1B1), a NADPH-dependent cytosolic oxidoreductase, plays a central role in the polyol pathway and is also crucial in hyperglycemia-induced tissue damage. In addition to its metabolic function, increased ALR2 expression has been reported in various malignancies, including hepatocellular and pulmonary carcinomas, highlighting the enzyme's potential as a therapeutic target at the metabolic-oncological interface. The aim of this study was to determine the inhibitory effects of various pyrimidine derivatives on aldose reductase enzyme and to evaluate the relationship between the structural properties of these compounds and their inhibitory activities. In this study, a series of seven pyrimidine and its derivatives (pyrimidine (1a), 4-amino-2-chloropyrimidine (1b), 4-amino-2,6-dichloropyrimidine (1c), 4-amino-5,6-dichloropyrimidine (1d), 4-amino-6-chloropyrimidine (1e), 4-amino-6-hydroxypyrimidine (1f) and 4-amino-2,6-dimethylpyrimidine (1g)) were systematically evaluated for their ALR2 inhibitory properties. Biochemical activities of pyrimidines and their derivatives against human ALR2 were profiled using a concise, chromatography-free route. Enzymatic inhibition tests revealed low μM KI values, with (1f) being the most potent (KI = 4.49 μM). It was also found to be the most potent inhibitor, surpassing the reference drug Epalrestat. In contrast, (1g), which contains methyl groups, was found to have the weakest inhibitory activity. The findings may contribute to the design of new pyrimidine-based inhibitors for the pharmacological control of diabetic complications.
Aldose reductase, pyrimidine, inhibition, enzyme
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia (high blood sugar). This disease, which is highly prevalent worldwide, is a major health problem that significantly negatively impacts public health. Hyperglycemia plays a significant role in the development of diabetes-related complications, such as nerve damage (neuropathy), eye damage (retinopathy), kidney disease (nephropathy), and cataracts. This is because, when blood sugar is high (hyperglycemia), more glucose than normal is diverted to the polyol pathway [1] (Figure 1).
Although considered a minor pathway in normal glucose metabolism, the polyol pathway becomes overactive under conditions of hyperglycemia, leading to osmotic stress and redox imbalance due to the formation of reactive oxygen species (ROS) [2]. Aldose reductase (ALR2; EC 1.1.1.21), a metabolic enzyme in the polyol pathway, is a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent enzyme that facilitates the reduction of diverse ketones and aldehydes to alcohols. [1]. In hyperglycemia, glucose is converted to sorbitol using the auxiliary cofactor nicotinamide adenine dinucleotide (NAD⁺). When NAD⁺ and NADPH levels in the cell decrease or are insufficient, excess sorbitol cannot be broken down and begins to accumulate within the cell. This buildup raises oxidative stress and throws off cells' osmotic equilibrium, which damages organs and cells and eventually results in consequences from diabetes. In addition, when sorbitol and its metabolites cannot be broken down sufficiently, they accumulate in the kidneys, retina and nerve tissue, contributing to the formation of diabetic complications [3].
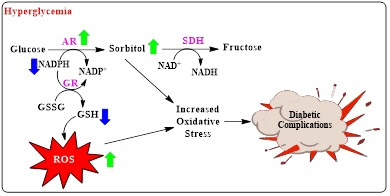
Figure 1. Polyol pathway mechanism in the formation of diabetic complications due to hyperglycemia
Aldose reductase (ALR2; EC 1.1.1.21) is a NADPH-dependent oxidoreductase enzyme that is part of the aldo-keto reductase (AKR) superfamily. The enzyme catalyzes the reduction of carbonyl compounds (e.g., aldehydes and ketones) to the corresponding alcohols. In humans, the ALR2 gene is called AKR1B1 and is located in chromosome 9q22.3. ALR2 is a protein of approximately 36 kDa, consisting of 315 amino acids. In its crystal structure, the enzyme exhibits a β/α-barrel (TIM-barrel) fold, and its active site contains the critical catalytic residues Tyr48, His110, and Asp43 [4].
It uses NADPH as a cofactor, which provides the electrons necessary for the reduction of the carbonyl group. Under normal physiological conditions, aldose reductase is involved in the detoxification of various endogenous aldehydes. It is known to be particularly active on sugars such as glucose, galactose, and ribose, as well as lipid peroxidation products (e.g., 4-hydroxy-nonenal). The enzyme is highly expressed in the kidney, liver, retina, nervous tissue, and lymphocytes. Furthermore, as an enzyme involved in the stress response, it reduces the accumulation of reactive carbonyl compounds in the cell [5].
ALR2 catalyzes the first and rate-limiting step of the polyol pathway in the pathogenesis of diabetic complications. In the polyol pathway, aldose reductase changes glucose into sorbitol, and then sorbitol dehydrogenase changes sorbitol into fructose. Under hyperglycemia, excess glucose is directed toward the polyol pathway; this process consumes NADPH and creates intracellular osmotic stress [2]. Decreased NADPH levels reduce glutathione reductase (GR) enzyme activity, leading to increased oxidative stress. Consequently, complications such as retinopathy, nephropathy, and neuropathy develop in the retina, kidney, and nerve tissues [3].
ALR2 is overexpressed not only in diabetic complications but also in many malignancies. Increased ALR2 expression has been reported, particularly in hepatocellular, prostate, and lung carcinomas. This increase is due to ALR2's influence on redox homeostasis and apoptotic signaling. Furthermore, ALR2 acts as a bridge between oxidative stress and inflammation by mediating the increase in inflammatory cytokines (e.g., TNF-α, IL-6) [6].
In recent years, various ALR2 inhibitors (ALR2Is) have been studied, including epalrestat [7], fidarestat [8], ranirestat [9], and sorbinil [10]. These molecules (Figure 2) exhibit pharmacokinetic performance with different levels of inhibitory activity. Also, their medicinal development has often been slowed down by problems including limited oral bioavailability, poor membrane permeability, metabolic instability, or lack of target selectivity [11].
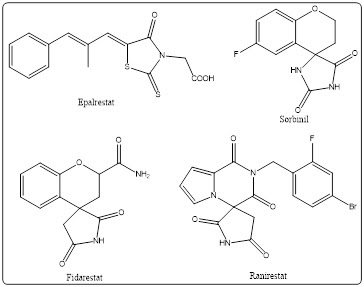
Figure 2. Chemical structures of some known ALR2 inhibitors
Heterocyclic compounds are of great importance in medicinal chemistry, agricultural chemicals, polymer materials, and various industrial products [12]. Nitrogen-containing heterocyclic compounds are abundant in nature and are also found in many natural products such as various alkaloids, vitamin derivatives, antibiotics, and hormones [13]. It is also known that approximately 60% of currently approved medical drugs contain heterocyclic nitrogen in their structures. Pyrimidine and its various analogs have long been a focus of interest due to their diverse biological activities. In addition to their medicinal properties, pyrimidine constitutes an important class of heterocyclic compounds because it is the basic molecule in the presence of the pyrimidine base, which consists of uracil, cytosine, and thymine, which form the structures of DNA and RNA [14].
Many chemicals are known to act on various enzymes. In fact, drugs exert their therapeutic effects by significantly altering the activities of enzymes. In this context, examining the effects of pyrimidine and some derivatives (Figure 3), known to have an impact on many metabolic diseases, on ALR2 activity is of critical importance. In summary, this study was designed to characterize novel ALR2 inhibitors and investigated the in vitro inhibitory effects of commercially available pyrimidines and some derivatives on human ALR2 enzyme activity.
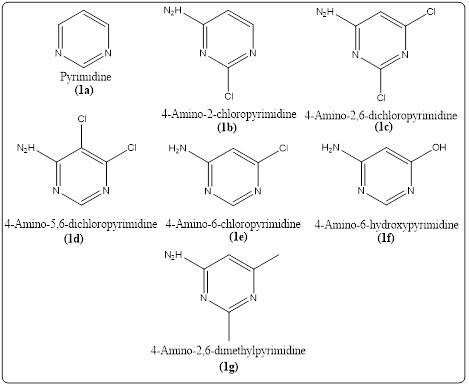
Figure 3. The pyrimidine and some derivatives used in the inhibition study
MATERIALS AND METHODS
All compounds used in the investigation were purchased from Sigma-Aldrich. The inhibitory profile of pyrimidine and its derivative compounds against ALR2 was examined using a cuvette-based spectrophotometric approach derived from the validated kinetic assay procedure by Cerelli [15] and all measurements were done in triplicate for consistency. NADPH oxidation at 340 nm (ε = 3.4 × 102 mM−1·cm−1) in a final assay volume of 1.0 mL was used to measure enzyme activity. Sodium phosphate buffer (10 mM, pH 7.4), NADPH (0.11 mM), and DL-glyceraldehyde as substrate at different concentrations (0.071–0.376 mM) made up the reaction mixture [16]. To achieve a final DMSO concentration of at least 1%, inhibitor stock solutions were made in DMSO and added. Before adding the substrate, the enzyme and inhibitors were preincubated on ice for one minute. All analyses were carried out for three minutes. The reference inhibitor was EPR. The kinetic mechanism of inhibition was identified using Lineweaver-Burk plots, and inhibition constants were computed as a result [17]. These plots were used to categorize inhibition types.
RESULTS
In this study, the effects of pyrimidine and some of its derivatives on ALR2 were investigated. The physiological inhibitory properties of pyrimidine and some of its derivatives against the ALR2 enzyme were analyzed in vitro and compared with the reference compound EPR. Inhibition data for pyrimidine and some of its derivatives (1a-g) are presented in Table 1.
Table 1. IC50 and KI values for EPR used as standard inhibitor with pyrimidine and its derivatives (1a–g) on ALR2
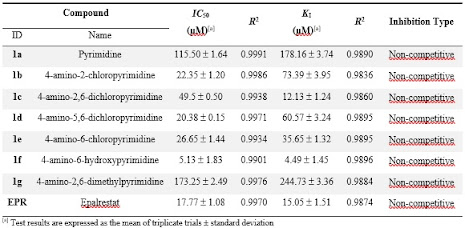
Regarding the inhibition of ALR2, it was observed that the analyzed pyrimidines (1a–g) exhibited inhibition with KI values ranging from 4.49 ± 1.45 μM to 244.73 ± 3.36 μM. In addition, compounds (1f) and (1c), which showed better inhibition than EPR, were found to be quite strong inhibitors compared to the clinically used reference agent EPR (15.05 ± 1.51 μM KI). While (1f) (KI value 4.49 ± 1.45 μM) was found to be the strongest agent, 4-amino-2,6-dimethylpyrimidine (1g) (KI value 244.73 ± 3.36 μM) was determined to have less effect against ALR2. The inhibitory activities of pyrimidine and some of its derivatives on ALR2 are as follows; 4-amino-6-hydroxypyrimidine (1f) > 4-amino-2,6-dichloropyrimidine (1c) > EPR > 4-amino-6-chloropyrimidine (1e) > 4-amino-5,6-dichloropyrimidine (1d) > 4-amino-2-chloropyrimidine (1b) > Pyrimidine (1a) > 4-amino-2,6-dimethylpyrimidine (1g).
IC50 and KI plots for EPR, used as standard inhibitors, with pyrimidine and its derivatives (1a–g) on the ALR2 enzyme are shown in figure 4 and figure 5.
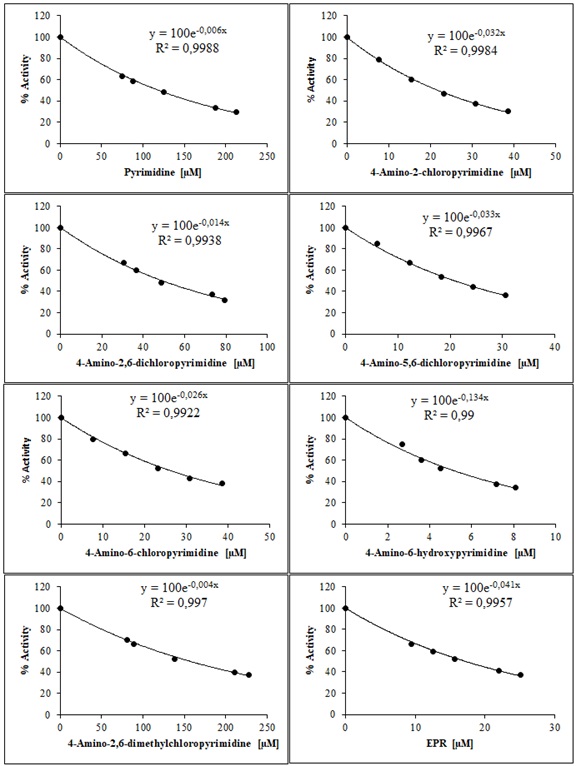
Figure 4. The IC50 graphs for EPR used as standard inhibitors with pyrimidine and its derivatives on the ALR2 enzyme
Figure 5. KI plots for EPR using pyrimidine and its derivatives (1a–1g) as standard inhibitors on the ALR2 enzyme
DISCUSSION
The central role of ALR2 in diabetic complications has made the enzyme a therapeutic target. Aldose reductase inhibitors (ARIs) developed for this purpose are classified into two main groups: Carboxylic acid derivatives: epalrestat, tolrestat, and zopolrestat. Spiron compounds and hydantoin derivatives: sorbinil, fidarestat, and ranirestat. Epalrestat is the only ALR2 inhibitor in clinical use in Japan and India [18]. However, most inhibitors have not achieved clinical success due to reasons such as low lipophilicity, poor bioavailability, or toxicity [16]. Current studies are investigating the efficacy of novel heterocyclic structures (phthalimide–benzoic acids, indoles, carbazoles, phenothiazines, ureido-benzenesulfonamides, benzotriazoles, and isoxazoles) in ALR2 inhibition [19-22]. Electron-withdrawing substituents (Cl, OH, NO₂) have generally been reported to increase inhibitory activity in these compounds [23].
The enhancement of inhibitory activity by chlorine and hydroxyl groups can also be explained by their potential to interact with polar amino acid residues (e.g., Tyr48, His110, Trp111, Cys298) in the enzyme's active site [4]. Previous molecular modeling studies have shown that these residues are important binding sites in the catalytic core of ALR2. In light of this information, the structural basis of the strong inhibitory activity obtained in our study may be associated with the high polarity and binding orientation, particularly in compounds containing hydroxyl groups.
This study evaluated the inhibitory effects of pyrimidine and its derivatives on the ALR2 enzyme. The findings indicate that pyrimidine derivatives have significant potential as ALR2 inhibitors. In particular, 4-amino-6-hydroxypyrimidine (1f) had the lowest KI value (4.49 μM) and exhibited stronger inhibition than the reference inhibitor EPR (KI = 15.05 μM). This result suggests that appropriate substitution of the pyrimidine skeleton can create high binding affinity.
In the literature, ALR2 inhibitors are reported to be considered promising therapeutic agents for the prevention of diabetic complications [2, 10]. In addition to clinical agents such as epalrestat, fidarestat, and ranirestat, new-generation heterocyclic compounds (e.g., thiazole, benzotriazole, pyrimidine, and indole derivatives) are being evaluated for enzymatic selectivity [8]. However, most existing inhibitors have not achieved clinical success due to limitations such as low bioavailability or pharmacokinetic instability [23]. In this context, the low-nanomolar inhibitors identified in this study, especially potent analogs such as compound (1f), are considered valuable for pharmacological development.
Another pharmacologically important aspect of pyrimidine derivatives is their biocompatibility and metabolic stability. Pyrimidine-based compounds have been reported in the literature to exhibit antioxidant, anticancer, antimicrobial, and antidiabetic properties [12]. Therefore, it is anticipated that pyrimidine derivatives may also exhibit metabolic regulatory effects beyond ALR2 inhibition.
When evaluated in terms of structure-activity relationship (SAR), it was determined that chlorine and hydroxyl groups increased inhibitory activity, while methyl groups decreased this effect. This may be due to the ability of electron-withdrawing groups (e.g., Cl, OH) to form stronger hydrogen bonds and van der Waals interactions with the enzyme's active site [24]. Similarly, studies conducted by Yapar and colleagues on hydrazone derivatives reported that electron-withdrawing groups (especially halogen and hydroxyl groups) enhanced ALR2 inhibition [1, 21]. This similarity suggests that the pyrimidine skeleton also benefits from similar electronic interactions.
Numerous studies have demonstrated that the ALR2 enzyme is inhibited by various substances. For instance, Papastavrou et al. [25] examined the inhibitory activity of novel compounds they synthesized by adding a trifluoroacetyl group to the pyrrole ring against ALR2 and demonstrated that all compounds were selective and potent ALR2 inhibitors. Maccari et al. [26] synthesized a series of new thiazolidinone derivatives as an ALR2 inhibitor and showed that these compounds were good ALR2 inhibitors. Additionally, Kim et al. [27] isolated 5-anthocyanins and 7-nonanthocyanins from the EtOH extract of Zea mays L. and demonstrated the inhibitory property of these compounds against ALR2.
Limitations of this study include the fact that only in vitro analyses were conducted and validation at the cellular level has not yet been performed. Similar studies conducted by Güleç et al. [28] detailed the binding modes after enzyme inhibition using molecular modeling, docking, and dynamic simulation analyses. Therefore, future studies are recommended to investigate the interaction mechanisms of pyrimidine derivatives with ALR2 using computational methods and to evaluate the stability and binding energies of enzyme-ligand complexes. Furthermore, testing the potent inhibitor compound (1f) in cellular oxidative stress and diabetic animal models will contribute to determining its potential therapeutic value.
CONCLUSION
This paper proposes a highly successful procedure for pyrimidines. Investigations were conducted on the inhibitory effects of pyrimidine and its derivatives on the activity of the ALR2 enzyme, which is recognized as a crucial target in the treatment of diabetes, cancer, and other conditions. In conclusion, this study demonstrates that pyrimidine derivatives can be potent and selective ALR2 inhibitors. In particular, the 4-amino-6-hydroxypyrimidine derivative (1f) exhibited inhibitory activity at low micromolar levels, surpassing the current reference inhibitor EPR. In particular, appropriate positioning of the chlorine and hydroxyl groups was found to enhance the interaction with the enzyme's active site, increasing inhibitory activity. The findings acquired from this research make an important contribution to the literature. We believe that the findings will contribute to both the design of new drugs to prevent diabetic complications and the discovery of new structural-biological correlations in the field of heterocyclic chemistry. The present research focuses on novel methods for measuring the course of diabetes mellitus by ALR2, which may lead to new and interesting options for target identification and medication discovery. Further biological research may benefit from the chemicals examined in the study and their inhibitory effects on the ALR2 enzyme.
Ethical Approval
This study does not involve any form of human and animal testing. Hence, the ethical approval is not required.
Conflict of Interest
No conflict of interest was declared by the authors.
Financial Disclosure
The authors declared that this case has received no financial support.
Author contributions
Ahmet Esat Göner and Hatice Esra DURAN have contributed equally to this study.





Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org