International Surgery Case Reports
OPEN ACCESS | Volume 8 - Issue 1 - 2026
ISSN No: 2836-2845 | Journal DOI: 10.61148/2836-2845/ISCR


Parvaneh Karimzadeh MD1,2, Zahra Hosseini nezhad MD2*, Mohammad Keramatipour PhD3, Abolfazl Heidari PhD4, Mitra Khalili MD5, Zahra babaei MSc2
1Pediatric Neurology Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2Pediatric Neurology Department, Mofid Children’s Hospital, Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
3Department of Medical Genetics, School of Medicine, Tehran, Iran University of Medical Sciences, Tehran, Iran
4Medical Geneticist, Reference Laboratory of Qazvin Medical University, Director Qazvin, Iran
5Pediatric Radiology, Mofid Childrens Hospital,Shahid Beheshti University of Medical Sciences,Tehran,Iran,
*Corresponding Author: Zahra Hoseini, Pediatric Neurology Department, Mofid Children’s Hospital, Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Received date: January 11, 2022
Accepted date: January 16, 2022
published date: Februaru 08, 2022
Citation: Karimzadeh P, Zahra H Nezhad, Keramatipour M, Heidari A, Khalili M, Babaei Z. (2022) “Adenylosuccinate lyase Deficiency (ADSL) and Report the First Case from Iran”. International Surgery Case Reports, 4(1). DOI: http;//doi.org/11.2022/1.1046.
Copyright: © 2022 Zahra Hosseini Nezhad. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Adenylosuccinate lyase deficiency is a neurometabolic disorder associated by accumulation of succinylpurines in body fluids that causes encephalopathy. It’s a rare neurological dysfunction with psychomotor retardation and epilepsy. We introduce here a five and a half year old patient who was referred to the Neurology Department, Mofid Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran. Chief complains were reported refractory seizure and neurodevelopmental delay (NDD).Patient was from Qazvin and was bedridden. He has been suffering from tonic seizure 3 to 11 times a day since he was 3 months. His parents were cousin. The patient had one older brother who had neurodevelopmental delay with seizure, too. He passed away at the age of 3.5 years old. ADSL deficiency has been documented by Whole exome sequencing (WES) in our patient and because of poor seizure control, Ketogenic diet (KD) was initiated and good results have been observed at the beginning steps.
Introduction
Adenylosuccinate lyase deficiency is a rare inherited neurometabolic disorder of purine metabolism with the combination of neurological and physiological symptoms such as psychomotor retardation, epilepsy, autistic behaviors, respiratory failure and Muscular hypotonia (1,2). For the first time in 1984, Jaeken and van den Berghe described it by finding succinylpurines in the cerebrospinal fluid (3).
ADSL deficiency is classified into three fatal to mild forms with wide spectrum of signs and symptoms. Patient's phenotypes have classified as severe form type I, milder type II and fatal neonatal form (4). The type I form is characterized by microcephaly, severe neurodevelopmental delay, seizures, and autistic features and most of the patients reported so far have ADSL type I (5). Type II form include moderate psychomotor delay and seizure and Fatal neonatal form instead is encephalopathy, Refractory seizure and respiratory failure and leads to early death (6).
The prevalence has been estimated 1 in 1.25 million and up to now less than 100 cases have been described in the world (7).
Here, we represent a five-and-a-half-year-old boy who referred to our neurometabolic clinic. He had been diagnosed as having ADSL deficiency with no obvious signs of disease progression and degradation.
Case Study
A five-and-a-half-year-old boy from Qazvin was transferred to Mofid Hospital, Shahid Beheshti University of Medical Sciences with Refractory seizure and Neurodevelopmental delay (NDD).
His parents were cousin and he was 4th child of a consanguineous marriage.
One of his older brother who was the second child in family suffered from NDD with refractory seizure and passed away at the age of 3.5 years old.
The patient was bedridden and has suffered from tonic seizure 3 to 10 times a day since 3 months of life.
He could have head control until being 3 months old and there after his general developmental condition seemed to progressively delayed; and now global developmental delay is obvious in physical and mental examination.
Different type of antiepileptic drugs had been prescribed due to seizure reduction, but without obvious control.
His neurological exam showed hypotonia, negative fix and follow with normal DTR and Magnetic resonance imaging (MRI) revealed evidence of Ventriculomegaly and Front temporal atrophy.
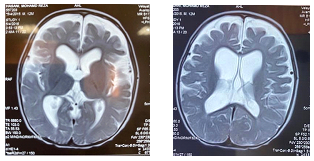
Hypomyelination, enlarged lateral ventricles and cortical atrophy
MRI shows delayed myelination, enlarged lateral ventricles, abnormal white matter signal, and volume loss throughout the cerebrum or cerebellum which is apparent in the first few months of life and radiologist may be the first individual to prompt this disease.
After neurological evaluation and laboratory investigations assessment, patient referred for genetic testing.
Genetic investigation
To investigate the genetic cause, whole blood samples were collected from patient and his parents in EDTA containing tubes. Genomic DNA was extracted from whole blood using Blood SV-mini kit (GeneAll Biotechnology Co., LTD, South Korea) according to the manufacturer instruction.
DNA sample quality control was performed before using DNA for whole exome sequencing. Quantity of DNA was measured by PicoGreen (Invitrogen, Thermo Fisher Scientific, USA) method using Victor3 fluorometry as well as a micro-spectrophotometer. Assessment of DNA condition was performed by gel electrophoresis.
Library preparation was performed using Agilent SureSelect Human All Exon V7 system (Agilent Technologies Inc.,Santa Clara, CA, USA) using manufacturer instruction. Sequencing of libraries was done by high-throughput paired-end sequencing using HiSeq2000 sequencing platform (Illumina Inc., CA, USA).
Sequencing short reads were aligned to the reference human genome hg19 from UCSC genome browser (University of California, Santa Cruz, USA) via the Burrows-Wheeler Aligner (BWA) program. Variant calling and filtering was done using Genome Analysis toolkit (GATK-v3.4.0). Detected variants were annotated using WANNOVAR software.
Proper filtering and then interpretation of a short list of variants in terms of pathogenicity was performed based on ACMG (American College of Medical Genetics and Genomics) guideline for variant interpretation.
To evaluate the pathogenicity of candidate variants, the potential impact of a given variant on the function or structure of the encoded protein was analyzed. The analysis was carried out based on conservation, physical properties of the amino acids or possible occurrence in regulatory or splicing motifs using bioinformatics tools. OMIM and PubMed was reviewed for previous publications related to the candidate causative gene.
After defining a pathogenic variant as the causative variant in the patient on WES data, patient’s homozygous genotype as well as carrier status (heterozygosity) of his parents was validated by PCR (Polymerase Chain Reaction) and Sanger sequencing using ABI 3500 Genetic Analyzer (Applied Biosystems Inc., CA, USA).
Results
Analysis of WES data ends up with a homozygous variant in exon 12 of ADSL gene on chromosome 22 (c.1277G>A, p.R426H, hg19: 22-40760969-G-A). Homozygosity of variant in patient was also confirmed by PCR-sanger sequencing (Figure 1-a). Parental analysis confirmed heterozygous carrier states in his parents (Figure 1-b-c) that is consistent with the autosomal recessive pattern of inheritance. Having these finding in addition to bioinformatic analysis, detected variant was classified as a pathogenic variant based on ACMG guideline for variant interpretation. Having this classification, diagnosis of disease is confirmed in our patient.
 Figure 1: Homozygosity for variant c.1277G>A in ADSL gene in patient (a) and heterozygosity in his mother (b) and his father(c) was confirmed by PCR-Sanger sequencing.
Figure 1: Homozygosity for variant c.1277G>A in ADSL gene in patient (a) and heterozygosity in his mother (b) and his father(c) was confirmed by PCR-Sanger sequencing.
In recent decades there was no effective treatment for adenylosuccinate lyase deficiency but currently, Ketogenic diet has mentioned as an effective treatment for ADSL deficiency(8).
Ketogenic diet was first developed in the early 1920s due to poor seizure control (9). It was high fat, adequate protein and low carbohydrate diet. Currently, there are different methods of KD that all have high fat and low carbohydrate components that can be administered after 48-72 hours of eating Ketocal formula by the gradual introduction of calories (10). Now, the Ketogenic diet is considered as a non pharmacological and effective treatment for GLUT-1 deficiency, Pyruvate dehydrogenase deficiency (PDHD), Refractory seizure and ADSL deficiency (11). There are various mechanisms that have been expressed for the antiepileptic effect of the ketogenic diet in ADSL deficiency.
Hypoglycemia decreased PH, ketone bodies and Acetone production, providing more gamma-aminobutyric acid (GABA) and purinenergic signaling hypothesis cause anticonvulsant actions of a ketogenic diet in ADSL deficiency patients (12).
In our patient, clinical trial with KD and dietary supplementation has begun and after diet initiation, frequency of seizures reduced significantly and it's important that systematic examinations and routine follow up testing have been checked for patient to prevent side effects that may present during the diet therapy.
Therefore, KD could be considered as a valid therapeutic option for ADSL deficiency patients with resistant seizures.
Discussion
ADSL deficiency is a rare neurological dysfunction disease in Iran, so its diagnosis is more complex because of insufficient data and reports. ADSL deficiency has been described by Jaeken and van den Berghe in 1984 for the first time (3,13) . From the first cases described in 1984 several cases of ADSL deficiency have been reported describing the clinical feature, laboratory data, treatment methods and disease complications.
Here we presented a five and a half year old boy with ADSL deficiency. He suffered from NDD with history of resistant epilepsy. For diagnosis, in addition to history, physical examination, Brain CT, MRI and WES were taken. Examinations showed hypotonia, negative fix and follow with normal DTR and MRI provided characteristic findings including: Ventriculomegaly and Frontotemporal atrophy.
Eventually, whole exome sequencing has documented Adenylosuccinate lyase deficiency.
There was no effective treatment for adenylosuccinate lyase deficiency but currently, Ketogenic diet has mentioned as an effective treatment for ADSL deficiency (14).
So classic ketogenic diet with 3:1 ratio and three-course meal has been prescribed with multivitamin, calcium and vitamin D supplementations.
At the beginning, patient's family wasn’t acquainted with this treatment method; they have been referred to nutrition department and diet therapy has been started with Ketocal formula. After 48 hours urine ketone body has been monitored, the test was positive (2+) and frequency of seizures has been changed. Currently, the episodes are very short and they appear one to two times per day.
Children on ketogenic diet therapy should have regular follow up visits after one month and then every 3months in the first year by pediatric neurologists and dietitians because there is a specific concern about side effects (15).
It's important to aware of side effects and any warning sign and symptoms such as lethargy, nausea, constipation and vomiting (16).
In our patient at the beginning and after 1 month of therapy the appetite reduced and he lost 2 kilo grams. So his mother complained about his child's constipation. Therefore, MCT oil has been added to his diet by dietitian to control the symptom of constipation
Acknowledgement
The authors appreciate all who supported this study
Author’s contribution
All authors have seen and approved the content of the final manuscript that is currently being submitted and have contributed significantly to the work.
Conflict of interest: None














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org