Pharmacy and Drug Innovations
OPEN ACCESS | Volume 4 - Issue 1 - 2025
ISSN No: 2994-7022 | Journal DOI: 10.61148/2994-7022/PDI

Ismail khan*, Abad Khan, Asmat Ullah, Lateef Ahmad, Waqar Ahmad
Department of Pharmacy, University of Swabi, Swabi-23340, Pakistan.
*Corresponding author: Ismail Khan, Department of Pharmacy, University of Swabi, Swabi-23340, Pakistan.
Received: November 04, 2021
Accepted: November 12, 2021
Published: November 16, 2021
Citation: Ismail khan, Abad Khan, Asmat Ullah, Lateef Ahmad Waqar Ahmad (2021). “Factors Affecting the Pharmacokinetics of Etoposide, Approaches to Enhance Bioavailability and Various Methodologies for its Analysis in Dosage Forms and Biological Fluids”. J Pharmacy and Drug Innovations, 3(1); DOI: http;//doi.org/03.2020/1.1037.
Copyright: © 2021 Ismail Khan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
DNA topoisomerase inhibitors have played a very significant role in the treatment of various malignancies like germ cell tumors, Kaposi’s sarcoma associated with AIDS, Hodgkin’s disease, Non-Hodgkin’s lymphoma, mycosis fungoides, acute myeloblastic leukemia, neuroblastoma, Ewing’s sarcoma, paediatric rhabdomyosarcoma, ovarian carcinoma, small cell lung cancers, non-small cell lung cancers, gastric cancer and hepatoma in the past two decades. These drugs are poorly water-soluble which necessitates including vehicles such as surfactants in their commercial formulations. Polyethylene glycol (PEG) and polysorbate-80 (Tween 80) dissolved in ethyl alcohol (EC) were used as co solvents/surfactants in the formulations, which are responsible for many of the unwanted effects such as hypersensitivity, hypotension and metabolic acidosis. These agents were also responsible to alter the pharmacokinetics and bio-distribution of these drugs. Also high doses, which need to be administered causes, the precipitation of these drugs, the surfactants in this case causes the infusion system to break. A number of strategies have also been adopted to develop surfactants free formulations, include GCPQ formulations and certain Glycol Chitosan formulations. An overview of all these formulations, their advantages & disadvantages and methods of analysis, their pharmaceutical & pharmacological approaches will be discussed in detail in this review.
1.Introduction
DNA topoisomerases are essential for DNA replication, transcription, chromosomal segregation and recombination. Two major topoisomerase forms are present in all cells: the type I enzyme and type II enzymes, which cut and pass double-stranded DNA. DNA topoisomerase I was first described in 1971[1-3] and DNA topoisomerase II in 1976 [4]. Several commercially available antineoplastic drugs are now known to be inhibitors of topoisomerase I (irinotecan, topotecan) or topoisomerase II (etoposide, teniposide, doxorubicin, daunorubicin, idarubicin, mitoxantrone).
Among the available drugs for chemotherapy, Topoisomerase Inhibitors are one of the best anti-cancer drugs that act by inhibiting topoisomerase enzymes, which catalyze the changes in DNA structure during cell cycle. It is believed that topoisomerase inhibitors damage the integrity of genome by blocking the ligation step during cells replication process and ultimately produce apoptosis and cell death [2, 5]. Generally, all cells have two types of topoisomerase enzymes, topoisomerase I enzyme first described in 1971 [6] which catalyzes cuts in single strand DNA and topoisomerase II enzymes which was first reported in 1976 [7] and interfere with double strand DNA.
Sub-families of Topoisomerases are;
There are so many sub-families of topoisomerases because different organisms posses different topoisomerase repertoires and similarly different regulatory strategies among different species.[8]
A number of chemotherapeutic drugs which are known to be inhibitors of topoisomerase enzymes are now available commercially including topoisomerase I inhibitors (camptothecin, topotecan, irinotecan) or topoisomerase II inhibitors (etoposide, teniposide, mitoxantrone, amsacrine, ellipticines, doxorubicin, daunorubicin, idarubicin, aurintricarboxylic acid) [9].
Synonyms of etoposide are;

Etoposide
Etoposide, a potent anti-neoplastic drug, was one of several podophyllotoxin derivatives that was first synthesized in the 1960s and was then introduced into cancer clinical trials in the early 1970s. It is a semi synthetic glucosidic derivative of podophyllotoxin, which is a natural product compound extracted from the dried roots and rhizomes of species of genus Podophyllin belonging to the family of Berberidaceae. Its chemical structure is shown in Figure 1. Etoposide provides superior pharmacological profiles and broader therapeutic potential compared to podophyllotoxin. Etoposide is a White to yellow-brown crystalline powder with empirical formula of C29H32O13 and molecular weight 588.56 g/mol. The melting point of etoposide is 236 - 251 °C. Etoposide is very much soluble in methyl alcohol and chloroform, slightly soluble in ethyl alcohol but only sparingly soluble in aqueous environment. Thus, drug formulations for intravenous administration contain organic co-solvents such as benzyl alcohol, polyethylene glycol, and ethanol [10-14].
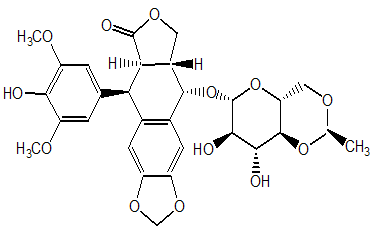
Figure 1: Chemical Formula of Etoposide
2.Pharmacology of etoposide
2.1 Mechanism of Action:
DNA topoisomerases are classified into two groups: topoisomerase I enzymes that induce single stranded cuts into DNA and topoisomerase II enzymes that cut and pass double stranded DNA. The major mechanism of action of etoposide is mainly explained by the interaction with topoisomerase II. By binding to topoisomerase II, etoposide stabilizes the cleavable complexes which results in double strand and single strand breaks in the DNA. Unlike podophyllotoxin, etoposide does not arrest cell division at the mitotic phase by binding to microtubules, but induces a premitotic block prevalent in the late S or early G2 period both in vitro and in vivo. Cells that are duplicating their DNA for the mitosis are very sensitive for this mechanism. The inability of etoposide to inhibit microtubule assembly is due to the glucoside moiety, which sterically blocks the interaction with tubulin. Breaks in the DNA are caused by either an interaction with DNA topoisomerase II or the formation of free radicals. Until now, it is still not clear how stabilization of the DNA topoisomerase II-etoposide complex ultimately leads to cell death and etoposide may also have other mechanisms of cytotoxicity [11-12, 15-16],[8].
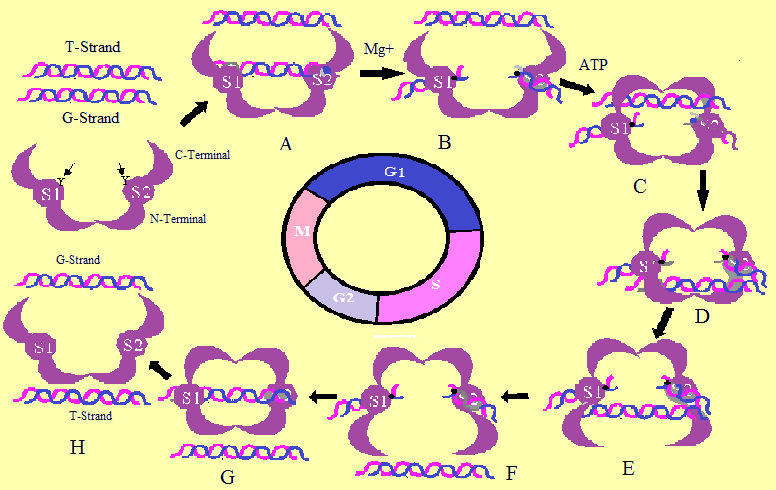
Topoisomerase II Enzymatic Action. The Topoisomerase II-poisons act by trapping the G-strand-enzyme intermediate in F step, thus, blocking relegation and enzyme release, leaving the DNA with a permanent double strand break. Where S, G1, G2, and M shows the phases of cell cycle.
2.2.Pharmacokinetics:
Oral etoposide has an average bioavailability of 50% (range, 17%-137%), with substantial intra-patient and inter-patient variability. Etoposide is widely distributed in the body and is highly bound to plasma proteins (greater than 95%). Approximately 50% (range, 20%-81%) of an etoposide dose is recovered in the urine as parent drug or glucuronide, with the remainder of the dose being unaccounted. The disposition of etoposide in patients with renal and hepatic dysfunction is discussed[17].
2.3.Indications
Etoposide is a highly effective and widely used agent for both the curative and palliative treatment of many neoplastic diseases including germ cell tumors, Kaposi’s sarcoma associated with AIDS, Hodgkin’s disease, Non-Hodgkin’s lymphoma, mycosis fungoides, acute myeloblastic leukemia, neuroblastoma, Ewing’s sarcoma, paediatric rhabdomyosarcoma, ovarian carcinoma, small cell lung cancers, non-small cell lung cancers, gastric cancer and hepatoma. Etoposide is also used in the preparatory chemotherapeutic regimens given prior to bone marrow transplantation in patients with advanced hematological malignancies [11, 13-15, 18-20].
2.4.Adverse effects
Low blood pressure, alopecia (loss of hair), pain/burning at the site if injection(I/V), constipation/diarrhea, metallic food taste, decreased white blood cells count, low red blood cells count (anemia), decreased platelet count, allergic reactions, nausea and vomiting, rash, fever, acute myeloid leukemia. Etoposide is known to cause fetal damage and birth defects[21].
2.5.Contraindications:
Etoposide is contraindicated in patients with known hypersensitivity to etoposide or to any component of the formulation (polysorbate 80), with severe myelosuppression, or with severe hepatic and/or renal impairment. Patients with low serum albumin may be at an increased risk of toxicity. Polysorbate 80 is associated with life threatening organ failure in preterm infants. Etoposide injection contains benzyl alcohol and should not be used in neonates. Etoposide has been shown to be embryotoxic, teratogenic, and oncogenic in animals and should not be used in pregnancy. Fertility may be affected. Present in breast milk, therefore breast feeding is not recommended.
3.Etoposide as Anti Neoplastic Agent
This review will concentrate primarily on the development of etoposide as an anti-neoplastic agent. Etoposide was the first agent recognized as a topoisomerase II inhibiting anticancer drug. Research on etoposide has helped the understanding of mechanisms by which drugs poison topoisomerase II. Recognition of the relationship of topoisomerase II inhibition and a resulting anti-neoplastic effect has stimulated development of other agents.
Currently available topoisomerase II inhibitors are briefly described. Etoposide is a semi synthetic derivative of podophyllotoxin, a compound extracted from the roots and rhizomes of the plants Podophyllum peltatum and Podophyllum emodi[22]. It is a cytotoxic drug and its mechanism of action is believed to be the inhibition of the topoisomerase II enzyme. Etoposide is widely used in chemotherapy for various solid tumors, including small cell lung carcinoma, testicular tumor, stomach cancer, ovarian cancer, and retinoblastoma[9].Since the aqueous solubility of etoposide is very low, this drug is commercialized in the form of non-aqueous parenteral solutions for intravenous use and oral soft gelatin capsules. However, both of these formulations have disadvantages. Etoposide precipitates from the parenteral solution when diluted with infusion fluids. In addition, cases of hypotension resulting from the rapid infusion of the drug and hypersensitivity reactions related to excipients of the formulation (ethanol, benzyl alcohol, polysorbate 80, and polyethylene glycol) have also been reported [23-24]. The oral administration of capsules containing a solution of etoposide in a solvent mixture exhibits low and variable bioavailability due, in part, to the inactivation of the drug in gastrointestinal fluids [24-25]
4.Formulations;
A number of etoposide formulations are available in market such as conventional oral, injectable and advanced nanoparticles formulations, which have certain merits and demerits; these are discussed below in detail;
4.1.Conventional Formulations of Etoposide
Due to hydrophobic and lipophilic nature, the clinical acceptable formulation of etoposide is challenging to date and the available commercial formulations therefore, contain various toxic adjuvant which have been reported to produce unwanted such as hypotension and metabolic acidosis [[26-27].
Moreover, current formulations are rapidly cleared from the blood circulation. Thus, requiring relatively high doses to be given to achieve sustained plasma levels. These suboptimal pharmacokinetics also lead to high peak doses more likely to induce myelosupression[28].The original formulation of etoposide, Vepesid® is formulated in a co-solvent mixture of polyethylene glycol (3250 mg), polysorbate 80 (400 mg) in ethyl alcohol and 100 mg of active drug etoposide (quantity sufficient to make 5ml) [29]. Common complications linked with adjuvant or co-solvent system of this formulation include hypotension and metabolic acidosis. These complications are believed to originate from polysorbate 80 (tween 80). Polysorbate 80 has been reported to provoke histamine release and produce hypotension in beagle dogs [29-30]. Polysorbate 80 has also been reported to alter the pharmacokinetics and bio-distribution of anti-neoplastic drugs such as doxorubicin [31-32] and anti-metabolite drug methotrexate [33] in experimental rodent models. Furthermore, during high-dose etoposide therapy where etoposide need to be administered in concentrated form, surfactants used in formulation has been reported to be the main cause of cracking of infusion system made of polyvinyl chloride plastic [34], unless proper safety measures are taken [34-35].
Precipitation of etoposide during dilution of etoposide injection is another issue associated with commercial formulation of etoposide. In order to get rid of precipitation, the concentration of etoposide in infusion fluids must not go beyond 0.4mgmL-1. Hence, large volume of I.V fluids (10-15L) are to be administered which further adds treatment related complications and poor patient compliance [36].
Bristol Myers Squibb developed an alternative formulation, Etophophos® that contains Prodrug, etoposide phosphate [37]. After I.V administration, both formulations produce similar pharmacological and toxicological results in comparative clinical trials [37-40]. Pharmacokinetic results indicate that Etophophos® at a standard dose produces equivalent serum concentrations to the original formulation, following either per oral [41-42] or I.V administration [38, 43]. It has been reported that Etoposide produces linear elimination pharmacokinetics when used in AHSCT at high dose [44], but with a three times higher Vd compared to standard-dose etoposide historical controls. This increase in volume of distribution might be due to perturbation of cell membrane permeability induced by polysorbate-80 [45].
4.2.Drawbacks of the current formulations:
Due to hydrophobic and lipophilic nature, the clinical acceptable formulation of etoposide is challenging to date and the available commercial formulations therefore, contain various toxic adjuvants which have been reported to produce unwanted side effects [26-27].Moreover, current formulations are rapidly cleared from the blood circulation. Thus requiring relatively high doses to be given to achieve sustained plasma levels. These suboptimal pharmacokinetics also lead to high peak doses more likely to induce myelosuppression [28-29].
Common complications linked with adjuvant or co-solvent system of this formulation include hypotension and metabolic acidosis[29-30]. Polysorbate 80 has also been reported to alter the pharmacokinetics and biodistribution of antineoplastic drugs such as doxorubicin [31-32] and antimetabolite drug methotrexate [33] in experimental rodent models. Furthermore, during high-dose etoposide therapy where etoposide need to be administered in concentrated form, surfactants used in formulation has been reported to be the main cause of cracking of infusion system made of polyvinyl chloride plastic [34], unless proper safety measures are taken [34-35].
Precipitation of etoposide during dilution of etoposide injection is another issue associated with commercial formulation of etoposide. In order to get rid of precipitation, the concentration of etoposide in infusion fluids must not go beyond 0.4mgmL-1. Hence, large volume of I.V fluids (10-15L) are to be administered which further adds treatment related complications and poor patient compliance [36],[45].
4.3.Alternative methods of formulations:
The most important objective of formulation development for etoposide is to get rid of the non-aqueous vehicle by reformulation etoposide in a well-tolerated vehicle which has the potential to improve the efficacy of etoposide based chemotherapy. Recently, much attention has been directed towards the development of aqueous based formulations for etoposide.
Some of these efforts to get a safer and better-tolerated formulation are parenteral emulsions [46], Tripalmitin Nanoparticles [47], PLGA nanoparticles [48], PLGA and PCL nanoparticles [49] , pH-sensitive strontium carbonate nanoparticles[50], Solid lipid nanoparticles [51],Parenteral Phospholipid-based Microemulsion [52], Positively charged Liposomes [53],Glyceride Lipids Nanoparticles [54], PLGA-MPEG and PLGA-Pluronic Block Copolymers Nanoparticles [55], Phospholipids complex [56], Cholesterol-modified carboxy-methylkonjac glucomannan conjugate [57], Erythrocyte ghosts [58], Microemulsion [59], Nano-crystalline suspensions[60], Carbon nanotube [61].
Among these, some dosage forms have been able to dissolve substantial amounts of etoposide and successfully improved the effects of anti-tumor activity in animal models.
Snehalatha et al. [49] encapsulated etoposide in PLGA and PCL nanoparticulate system. In vivo results in experimental rodents’ models demonstrated increased bioavailability and reduced toxicity. Similarly, Yadav and Krutika [62] prepared and optimized sustained release PLGA nanoparticle formulation of etoposide by double factorial design for continuous I.V administration. From the study results, they concluded that PLGA based etoposide nanoparticles showed enhanced stability in terms of particle size and drug content for relatively extended time. Wang and his co-workers loaded etoposide in star copolymer of poly (ε-caprolactone) and PEG. Etoposide was successfully loaded to a level of 22% (w/w) using star copolymer. Moreover, from invitro cytotoxicity study, they concluded that the star-PCL-PEG copolymer was nontoxic [63].
Yadav and Chuttani[55] synthesized PLGA-mPEG and PLGA-PLURONIC copolymers and were loaded with etoposide by high pressure homogenization. Blood clearance studies in rat showed that high concentration of 99mTc labelled nanoparticles were present in systemic circulation compared to free etoposide. Biodistribution results in experimental animal model (mice) confirmed reduced uptake of prepared nanoparticles by liver and spleen (RES) due PEG that impart steric properties to the nanoparticles. Similarly, Wu et al [56] have shown that the phospholipid complex self-emulsifying drug delivery system (PC-SEDDS) enhanced the oral bioavailability of etoposide. Sengupta et al.[53]have reported that Encapsulation of etoposide in liposomes increased the bioavailability and half-life of etoposide to a level of 1.7 fold and 3.1 fold, respectively.
Reddy Et al, prepared tripalmitin nanoparticles incorporating the anti-cancer drug etoposide by melt emulsification and high-pressure homogenization, followed by spray drying of the nano-dispersed material. The resulting nanoparticles possessed either a negative (ETN) or a positive (ETP) charge. Radiolabeled etoposide nanoparticles of both types were injected into mice; the ETP nanoparticles produced a relatively high distribution in the bone and the brain (14-fold that of etoposide alone) 4 hours post injection, which was much better than that by the ETN nanoparticles. The ETP nanoparticles possessed a long-circulating property and their effectiveness for targeting drugs both to tumors and to the brain should be beneficial.
However, problems have still been encountered with these alternative methods, such as in vivo stability of liposomes and dosage limiting toxicity of the dosage forms used [64-65]. The possible leakage of the drug from the liposomes formulation constrained the successful development of formulation using liposome, which is a promising drug carrier. The mixed micelles formulation and parenteral emulsions have the similar safety problems to be resolved. New formulations with more consistent release properties are necessary to be developed for successful delivery of etoposide to human body.
None of the methods seemed to be effective enough to replace non-aqueous based vehicle although the various approaches used so far have shown a lot of promise for etoposide delivery. The final product for human use is still far away. For that reason, much attention has been paid to discover well tolerable carriers in order to get better efficacy of etoposide in clinical chemotherapy.
Of these alternatives solutions, polymeric nanoparticles and polymeric micelles gained much attention and importance owing to their ability to accumulate in inflamed areas of the body [66-67]. Moreover, they possess better stability in biological fluids and during storage. Clinical trials showed satisfactory results [68-69] and thus, the use of nanoparticles of biodegradable polymers for chemo-embolization has been pursued in efforts to achieve the desired result of enhancing the therapeutic efficacy of anticancer agents while minimizing its systemic order effects.
Nanoparticles of biodegradable polymers represent a chemotherapeutic engineering solution or cancer nanotechnology solution. Nanoparticles composed of biocompatible and biodegradable polymers improved the in vivo characteristics including pharmacokinetics and biodistribution and hence chemotherapeutic effects of formulated drugs [70-71]. Results from a number of investigations have confirmed the increased accumulation of nanoparticulate in solid tumors due to enhanced permeability and retention (EPR) effect [72-73]. Similarly, it was demonstrated that nanoparticles formulation could overcome the MDR mediated by P-gp and thus increase the level of drug inside the cancer cells [74-76].
4.4.The impact of vehicles on efficacy:
Some co-solvents are used in the formulations such as polyethylene glycol, tween-80 and ethyl alcohol, which cause complications. Moreover, current formulations are rapidly cleared from the blood circulation. Thus requiring relatively high doses to be given to achieve sustained plasma levels. These suboptimal pharmacokinetics also lead to high peak doses more likely to induce myelosuppression[28].
The original formulation of etoposide, Vepesid® is formulated in a co-solvent mixture of polyethylene glycol (3250 mg), polysorbate 80 (400 mg) in ethyl alcohol (quantity sufficient to make 5ml) per 100 mg of active drug etoposide [29]. Common complications linked with adjuvant or co-solvent system of this formulation include hypotension and metabolic acidosis. These complications are believed to originate from polysorbate 80 (tween 80). Polysorbate 80 has been reported to provoke histamine release and produce hypotension in beagle dogs [29-30]. Polysorbate 80 has also been reported to alter the pharmacokinetics and biodistribution of antineoplastic drugs such as doxorubicin [31-32] and antimetabolite drug methotrexate [33] in experimental rodent models. Furthermore, during high-dose etoposide therapy where etoposide need to be administered in concentrated form, surfactants used in formulation has been reported to be the main cause of cracking of infusion system made of polyvinyl chloride plastic [34], unless proper safety measures are taken [34-35].
Precipitation of etoposide during dilution of etoposide injection is another issue associated with commercial formulation of etoposide. In order to get rid of precipitation, the concentration of etoposide in infusion fluids must not go beyond 0.4mgmL-1. Hence, large volume of I.V fluids (10-15L) are to be administered which further adds treatment related complications and poor patient compliance [36].
5.Method of Analysis of Etoposide
The clinical pharmacokinetics of etoposide has been reported. Most of published studies on etoposide have reported a two compartment pharmacokinetics of etoposide with shorter half-life of ~ 6–8 h [77]. However, in some cases, a three compartmental pharmacokinetics with longer half-life of 20–46 h has been reported [78]. Due to its multi-compartmental pharmacokinetics and larger individual variations, a sophisticated analytical method is required.
A number of analytical techniques using HPLC [22, 79-82], LC-MS [83] and UPLC-MS [84] have been reported for quantification of etoposide in biological samples. Most of the reported HPLC methods used for quantification of etoposide during pharmacokinetics studies have used electrochemical detectors [79, 81-82]. However, the shortcomings of using electrochemical detectors are their adjustment and the frequent cleaning of the working electrode as they are contaminated during use. Furthermore, the long run time due to late eluting peaks in biological samples further adds complications to the previously described methods. The only sensitive HPLC-UV method used for the quantification of etoposide in plasma is that reported by Shirazi et al [22]. However, the limitation of this method is the column high temperature (600c). furthermore, all the cited HPLC methods have used complex extraction procedures [85-87], complicated systems gradient systems [86], longer analysis time [86, 88]making them unsuitable for routine analysis.
Besides sensitive LC-MS and UPLC-MS methods, which due to high cost and special requirements are not available in the majority laboratories, the reported methods have been designed for a large volume of biological samples and limit their use for determination of etoposide in small biological samples. In majority of pharmacokinetics and tissue distribution studies, animals (rodent) models are usually used as human surrogates, but the limitation of such studies is the minute biological sample sizes. Thus the key considerations which should be consider during the development of analytical method for performing pharmacokinetic studies in animals for which serial biological samples are collected are the analytical sensitivity and samples volumes.
|
Mobile phase |
Stationary Phase |
Flow rate |
Internal standard |
Extraction procedure |
Detection |
|
Methanol, distilled water, and acetonitrile (55:42:3,v/v/v) |
C18;150 × 6.0 mm x5μm |
1.5ml/min, 40°C |
diphenylhydantoin |
dichloromethane |
SPD-6A UV detector 229 nm[89] |
|
methanol, acetonitrile and monopotassium phosphate (0.020 M) (18:19:63, v/v/v) pH 5.2 |
Chromolith RP-18 (100 mm×4.6 mmx5 μm) |
0.5ml/min |
Lamotrigine |
chloroform and n-hexane (80:20, v/v) |
DAD UV/VIS detector 285 nm[90] |
|
acetonitrile and acetic acid 4% (70:30) |
C18 (250 x 4.6 mm; 5 µm), at 25 °C |
2 ml/min |
|
|
(DAD) UV detection at 285 nm |
|
methanol: water (1:1, v/v) pH was maintained at 4.20 with 0.2N HCl |
C18 ODS Hypersil (250 × 4.6 mm x5 µm) |
1 ml /min |
Nefopam |
|
UV / Vis detector at 204 nm[91] |
|
water, methanol and acetic acid in the ratio of 45 : 54 : 1 |
RP C18 |
1.5 ml/min
|
Phenytoin |
chloroform |
254 nm [53] |
|
acetonitrile- water-glacial acetic acid (35:64:1, v/v) |
Bondapak phenyl analytical column
|
1.0 ml/ min |
Teniposide |
chloroform |
fluorescence detector, Fluorimetric detection [80] |
|
Methanol: water (45:55 v/v) |
Bondapac C18 column
60°C |
2.8 ml/min |
Phenacetin |
Ether |
SPD-6AV 220 nm[22] |
|
methanol/water/acetic acid (54:45:1) |
Phenomenex C18 column (250 mm,3.6 mm x5-µm |
1 ml/min
|
Phenytoin sodium |
chloroform |
PDA 254 nm[46] |
|
acetonitrile and water (35:65, v/v) Brain sample water/aceticacid/methanol (54:1:45) |
Phenomenex C18 column (size 100 x 4.6 mm,x 5µ. 45° C |
1.5 ml/min |
Teniposide |
acetonitrile and chloroform |
UV method 254 nm[92] |
|
water-acetonitrile-acetic acid (690:300:10, v/v/v)
|
Bondapak phenyl analytical column (300 mm x 3.9 mmx0 µm; |
1ml/min |
Teniposide |
1,2-dichloro ethane
|
281 nm wavelength Detector (TSP)[93]
|
|
methanol: water (60:40, v/v) |
C18- µBondapak column 30 cm x 4.0 mmx10; |
1 ml/min. |
Teniposide |
Chloroform |
wavelength detector set at 254 nm[44] |
|
acetonitrile-acetic acid-water (34:1:65), pH 4.0 |
C 18 ( 6.35 mm and length 25 cm) |
1.5 ml/min |
|
|
UV detector 230 nm[94] |
|
methanol/water/acetic acid (60:39:1) |
Nucleosil@ Phenyl column 7 µm (25 cm) |
1.0 ml/min, |
Teniposide |
chloroform |
UV- at 280 nm
[95] |
|
35% acetonitrile, 64% water, and 1% acetic acid |
µBondapak C18 phenyl column (10 mm×3.9 mm×300 mm |
1.5 ml/min |
Teniposide |
40% zinc sulfate |
PDA 230 nm[96] |
|
Methanol: Water (55 : 45) |
reversed phase YWG-C18 column |
|
Teniposide |
ethyl acetate |
UV detection at 254 nm[97] |
|
Methanol: Water (55 : 45) |
C18-reverse phase column |
1.2 ml/min, |
|
|
absorbance at 254 nm[98] |
|
Methanol: Water(70:30) |
C18 column at 30o C. |
0.7ml/min |
|
|
detected at 285nm |
|
Methanol: Water (55 : 45) |
150, 4.6 mm x 5 µm |
1.0 ml/min, |
|
|
UV 228nm[99] |
Table 1: HPLC Method of analysis for Etoposide
6. Approaches to enhance the bioavailability of etoposide:
6.1.Pharmaceutical Approaches:
Several pharmaceutical approaches were adopted in order to overcome the problems of oral solubility and erratic bioavailability. These are
6.1.1.Prodrug
An etoposide phosphate has been developed as a water-soluble pro-drug for clinical use. In patients with established solid tumors, the bioequivalence of etoposide phosphate to etoposide has been demonstrated. However, variable conversions of etoposide phosphate to etoposide within the intestinal lumen after oral administration remained a major cause for high inter-patient pharmacokinetic variability. Variability in absorption of etoposide is considered to play an important role in its instability in gastric or intestinal solutions [100-101]; however, some earlier approaches using drugs that influenced the rate of gastric emptying while modulating the time of drug absorption did not significantly alter the etoposide AUC or bioavailability[102]. Attempts to enhance the aqueous solubility and dissolution rate of etoposide were made by preparing various polymorphs of etoposide by crystallizing it from organic solvents [103].
6.1.2.Microemulsions
Several drug-targeting vehicles such as phospholipid-based microemulsions and cholesterol-rich microemulsions of etoposide have been found to be robust, safe and suitable for patient use [52, 104].
6.1.3.Micelles
Amphiphilic poly(2-oxazoline)s micelles have been developed as a promising high-capacity delivery platform; etoposide solubilized in defined polymeric micelles were found to achieve high total loading capacities [105]. Fatty acid chain length grafted to etoposide delivery has been another approach; etoposide showed high solubility in methoxy polyethylene glycol (MPEG) micelles [106]. Di-block copolymers of MPEG-b poly (ε-caprolactone) of six different molecular weights were used for fabrication of etoposide-loaded micelles by nanoprecipitation technique. Compared with plain etoposide, these miceller formulations have shown enhanced permeability and retention effect in Ehrlich ascites tumor-bearing Balb/C mice. Similarly, Tyr-Ile-Gly-Ser-Arg-conjugated etoposide loaded micelles were shown to enhance cytotoxicity and cellular uptake with significant reduction in colony formation and cell migration activities compared with non-conjugated micelles in over expressed tumor cells [107]. Other miceller formulations included MPEG poly epsilon caprolactone and were found efficient as drug delivery vehicles for pancreatic cancer therapy [108]. An etoposide-loaded linear PEGylated as well as PEG-b-poly (D, L lactic acid) offered a promising alternative for combination drug therapy without formulation related side-effects [109]. Etoposide encapsulated in the micelles formed from poly(epsilon-caprolactone)-poly(ethylene glycol) and poly(Lactide), poly(ethylene glycol) exhibited high etoposide loading capacity and were found suitable as a potential drug delivery carrier [110]. Enhancement of etoposide uptake by tumor via subcutaneous injection through etoposide-loaded polysorbate20 micelles resulted in significantly higher tumor uptake and prolonged tumor retention due to relatively high brain concentrations compared with etoposide [111]. Micelles containing poly (N-vinylpyrrolidone)-block-poly(D,L-lactide) were also found to be efficient solubilizers of teniposide and etoposide [112].
6.1.4.Nanoparticles
High residence of nanoparticles, compared with etoposide, was suggested to be advantageous as drug carriers for etoposide in enhancing the bioavailability and reducing the etoposide associated toxicity [49].Etoposide loaded into strontium carbonate nanoparticles, a novel biodegradable nanosystem, possessed both high loading capacity and efficient encapsulation, and were more potent in antitumor activity as compared with free etoposide [113]. Use of poly (lactide-co-glycolide)(PLGA) and PLGA/P188-blended nano-encapsulations over pre-existing etoposide formulation induced improved cytotoxic activity, showing a promising perspective for parenteral delivery of etoposide [114]. Recently, PLGA-PEG nano-carriers have been considered to be a better administration schedule in multiple drug delivery in cancer chemotherapy[115]. In a recent study colloidal formulations based on poly(butyl cyanoacrylate) nanoparticles of etoposide using two different non-ionic colloidal stabilizers (pluronic-F68and polysorbate 80) exhibited the highest cytotoxicity towards adenocarcinoma human epithelial (A549) cells[116]. Etoposide and rubusoside (RUB) nanoparticles completely reconstitutable in water and remained stable for at least 24 h. RUB has been shown to effectively solubilize and stabilize etoposide [117]. Etoposide-loaded nanoparticles were also prepared using PLGA, PLGA-MPEG block copolymer and PLGA-Pluronic copolymer. PLGA-based nanoparticles showed higher cell uptake and cytotoxicity compared with that of the free drug [118]. Etoposide-loaded and folic-acid-attached polymer poly(3-hydroxybutyrate-co-3- hydroxyl hexanoate (PHBHHX) nanoparticles were more effective on HeLa cells than etoposide-loaded PHBHHX nanoparticles without folic acid [57]. Etoposide nano-structured lipid carrier formulation remarkably improved the oral bioavailability of etoposide phosphate [99]. Intravenous administration of etoposide-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles of sizes 105 nm and 160 nm in mice and rats were present in the blood up to 24 h at higher levels than that of pure drug [119]. A sustained release formulation of etoposide-loaded biodegradable nanoparticles has been developed to replace the conventional therapy of continuous intravenous administration. Studies showed that the etoposide-loaded PLGA nanoparticles sustained the release up to 72 h [119]. Functional nano-materials that included gold, silver nanoparticles and single wall carbon nanotubes were delivered to the two cell lines, MLO-Y4 osteocytic cells and HeLa cervical cancer cells, in combination with etoposide, showed higher efficacy. Etoposide loaded with tripalmitin, glycerol mono-stearate and glycerol di-stearate nanoparticles showed greater and prolonged apoptotic induction properties, resulting in the higher increase in survival time of tumor-bearing mice compared with free etoposide [120]. Pharmacokinetic data of etoposide incorporated tripalmitin nanoparticles radiolabeled with Technetium-99m revealed high blood concentrations and prolonged blood residence time. In another study, etoposide lipid nano-capsules showed higher efficiency than the drug solution in glioma cell lines [121]. Etoposide loaded nanoparticles with glyceride lipids characterized and evaluated for in vitro steric stability and drug release characteristics [122]. Recently an HPLC method with fluorescence detection was developed and fully validated for determination and pharmacokinetic study of etoposide-loaded nanoparticles in dog plasma after intravenous administration [123].
6.1.5.Liposomes
Encapsulation of etoposide in lipid nanospheres (LN) improved the anticancer activity, and further inclusion of polyethyleneglycol-distearoyl phosphatidyl ethanolamine (DSPE-PEG) increased the circulation time and stability of LN. Folate-targeted etoposide-encapsulated lipid nanospheres showed higher tissue distribution of the drug in the kidney of normal mice compared with that of non-targeted etoposide or a commercial formulation. Etoposide lipid nanocapsules showed 4- to 40-fold higher efficiency than the drug solution [124]. Pulmonary liposomal delivery of etoposide showed better particle fraction and drug content over the period of 6 months[125].Liposomal etoposide were found to enhance the cytotoxicity when used alone or in combination with p53 tumor suppressor gene in non-small-cell lung cancer cell lines. These formulations when developed as dry powder inhaler showed significant in vitro lung deposition pattern and demonstrated new strategy to treat resistant lung cancer [126]. Liposomal etoposide have shown an improved pharmacokinetic profile: 60% increase in AUC with a 35% decrease in clearance, resulting in 70% increase in the mean residence time of the drug [85]. Liposomized etoposide and tuftsin-bearing liposomized etoposide formulations were found to reduce tumor volume and tumor growth, and was considered a promising treatment strategy for various forms of cancers, including fibrosarcoma [127]. Similar anti-metastatic activity of etoposide liposomes was also observed against pulmonary tumor nodule formation in murine experimental B16F10melanoma model [128]. Encapsulated etoposide in cationic liposomes significantly delayed tumor growth and were found to increase the area under the concentration vs time curve and half-life [53].
6.2.Pharmacological approach
Equally, noteworthy developments are documented for natural compounds from medicinal plants, which have been evaluated in order to explore bioavailability enhancement of etoposide. Application of P-glycoprotein (P-gp)/CYP3A4 dual role inhibitors in improving per oral drug delivery have gained special interest. P-gp expressed in the apical membranes of the epithelial cells of the intestine is known to reduce the oral bioavailability of a wide range of drugs, including etoposide. Etoposide is degraded via complex metabolic pathways.CYP3A4 is a principal isoform involved in the 3′-demethylationof etoposide, with the suggestion that CYP1A2 and 2E1are the minor isoforms involved in the etoposide metabolism[129]. Therefore, the possibility of improving the bioavailability of etoposide by combining this anti-cancer agent with several pharmacologically active substances, especially P-gp/CYP3A inhibitors, has been explored in recent times. Several known P-gp inhibitors have been shown toincrease the bioavailability of etoposide by reducing its efflux from target sites. Eudesmin a (bicyclic lignin) and diphyllin (aryl naphthalene lignin) isolated from H. perforatum Kar et Kir, ( Rutaceae) reversed P-gp mediated multidrug resistance(MDR) in MDR1 transfected Madin-Darby canine kidney(MDCK-MDR1) and doxorubicin-resistant human breast carcinoma cells (MCF7/Dox) [130]. Quercetin, a flavonoid with P-gp modulating activity, has been reported to increase etoposide absorption in everted sacs of rat jejunum or ileum [131]. A similar effect was also noticed with quinidine, an anti-arrhythmic agent in the rat everted gut sacs; its intravenous perfusion increased the serum level of etoposide in a dose-dependent manner [132]. Ketoconazole increased the area under the plasma concentration–time curve (AUC) of oral etoposide by a median of 20%. Ketoconazole reduced the apparent clearance of oral etoposide, did not alter its toxicity profile and did not reduce inter-patient pharmacokinetic variability [133]. Ketoconazole increased systemic exposure of etoposide due to inhibition of hepatic CYP3A4 [134]. Curcumin was found to increase the oral bioavailability (AUC and Cmax) of etoposide, possible due to inhibition of the P-gp efflux pump in the small intestine and possibly by reduced first-pass metabolism in the small intestine by inhibition ofCYP3A activity in rats. N-octyl-O-sulfate chitosan (NOSC) and verapamil enhanced the transport of etoposide from apical side to basolateral side in Caco-2 cell monolayers. Moreover, both these agents decreased the transport of etoposide from basolateral side to apical side, by inhibiting P-gp[135]. Orally administered morin (an inhibitor of CYP isozyme and P-gp) significantly increased the AUC, Cmax and the absolute bioavailability of orally administered etoposide [136]. Kaempferol also enhanced the AUC of intravenously administered etoposide due to inhibition of Cytochrome P450 CYP3A and P-gp[137]. Quercetin, a P-gp and CYP3A inhibitor, altered the pharmacokinetic parameters of etoposide in the orally treated group, but not in the intravenous treated group. Quercetin significantly increased the AUC and absolute bioavailability of orally administered etoposide and decreased the total body clearance (CL) of oral etoposide mainly due to inhibition of P-gp-mediated efflux and CYP3A catalysed metabolism in the intestine [137]. Potentiation effect of wogonin, a flavone in the roots of Scutellaria baicalensis, was observed to potentiate the anticancer action of etoposide due to P-gp inhibition and accumulation of this agent in etoposide-induced apoptosis in tumor cells [138-139]. A piperine analogue, namely,4-ethyl 5-(3,4-methylenedioxyphenyl)-2E, 4E-pentadienoicacid piperidide (PA-1), was shown to cause 2.32-fold enhancement of the absolute bioavailability of co-dosed etoposide in mice [84]. Enhancement in the oral bioavailability of etoposide by PA-1 could possibly be due to its ability to modify P-gp/CYP 3A4-mediated drug disposition mechanisms [140].
7.Conclusion
Several pharmacokinetic and biopharmaceutical aspects have been suggested to play a major role in the poor/variable oral bioavailability of etoposide, such as its poor dissolution characteristics, rapid elimination via P-glycoprotein. In the last two decades, many novel approaches have been explored in order to overcome these limitations, which have been discussed.
We are thankful to the University of Peshawar for providing the research facilities.

Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org