Pharmacy and Drug Innovations
OPEN ACCESS | Volume 4 - Issue 1 - 2025
ISSN No: 2994-7022 | Journal DOI: 10.61148/2994-7022/PDI

Sunil Chaudhry1*, Avisek Dutta2
1Honorary Medical Director, Bioclinitech Technologies Pvt Ltd Mumbai and GPAT tutor.com
2Service Delivery Manager, Cognizant Technology Solutions Pvt Ltd, Kolkata
*Corresponding author: Sunil Chaudhry, Honorary Medical Director, Bioclinitech Technologies Pvt Ltd Mumbai and GPAT tutor.com
Received: April 20, 2021
Accepted: May 10, 2021
Published: May 13, 2021
Citation: Sunil Chaudhry, Avisek Dutta. “Managing Jeopardy of Pulmonary Hypertension.” J Pharmacy and Drug Innovations, 2(4); DOI: http;//doi.org/03.2020/1.1017.
Copyright: © 2021 Sunil Chaudhry. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Idiopathic pulmonary hypertension, for which the cause is unknown, is more common in young adults and is approximately twice as common in women as in men. One third of patients with left heart failure have pulmonary hypertension. The annual mortality rate for pulmonary hypertension is 15%. Therapeutic options for the patient with PAH continue to expand through basic discovery and controlled clinical trials. Recent advancements in PAH-are targeted therapies and interventional-surgical procedures which contribute to improvement in quality of life and survival.
Introduction: The term PH means high blood pressure in the lungs. Pulmonary arterial hypertension (PAH) is clinically characterized by increasing pulmonary arterial pressure in the absence of elevated left heart pressure. If untreated, PAH leads to right ventricular failure, volume overload, and death Hypoxia is well-known as a risk factor for PH. Brain-derived neurotrophic factor (BDNF) regulates endothelial nitric oxide synthase (eNOS) and thus modulates pulmonary vasodilation shown in Figure 1
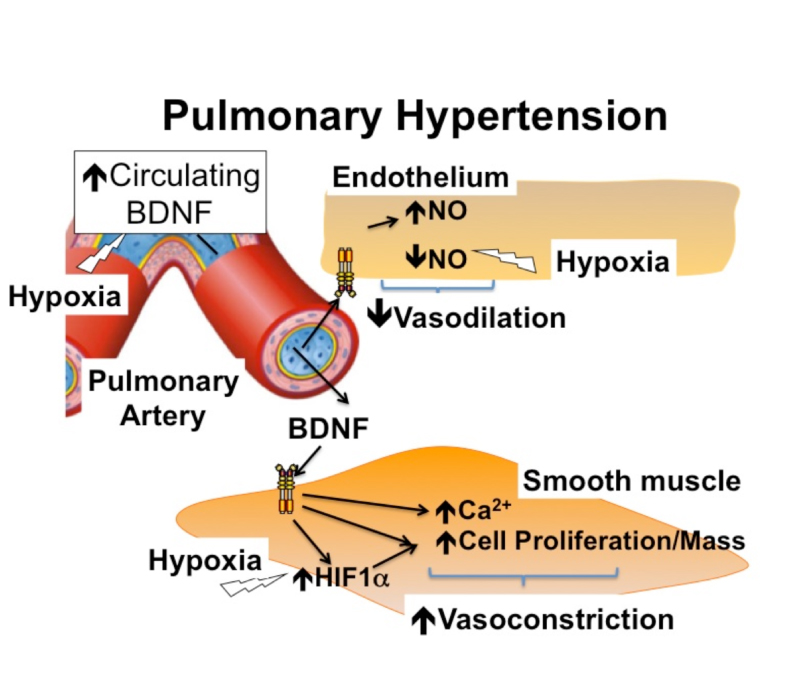
Figure 1
PAH is defined by a mean pulmonary artery pressure at rest > or =25 mm Hg in the presence of a pulmonary capillary wedge pressure < or =15 mm Hg. Normal pulmonary arterial pressure in healthy adult is 12-16mmHg and a normal wedge pressure 0f 6-12mmg Hg [1] Present estimates suggest a pulmonary hypertension prevalence of about 1% of the global population, which increases up to 10% in individuals aged more than 65 years. Pulmonary arterial hypertension, especially the idiopathic form, although still a rare disease with an incidence of 2–5 per million adults, is increasingly being diagnosed in elderly people [2].
|
Group |
Definition |
Selected Ethology |
|
Group 1 |
Pulmonary arterial hypertension (PAH) |
Heritable PAH Idiopathic PAH Congenital heart disease & PAH Connective tissue disease & PAH |
|
Group 2 |
PH due to Left heart disease |
Left ventricular systolic and diastolic dysfunction Mitral Valvular heart disease |
|
Group 3 |
PH due to Lung Disease and or hypoxia |
Chronic obstructive pulmonary disease Interstitial lung disease Developmental lung disease |
|
Group 4 |
Chronic thromboembolic PH |
|
|
Group 5 |
PH with multifactorial modes |
Sarcoidosis , Chronic haemolytic anaemia |
Clinical Classification of Pulmonary Hypertension - Table 1
[3]
The most common overall causes of pulmonary hypertension are
It can take months, even years, before the constrictions and narrowing in the arteries become severe enough that noticeable pressure begins to build. Pulmonary hypertension symptoms include Dyspnea, initially while exercising and eventually while at rest, Fatigue, Dizziness or fainting spells (syncope), Chest pressure or pain, edema in ankles, legs and eventually ascites, and cyanosis. The pulse is racing [4].
Mechanisms of Pulmonary Hypertension (shown in Figure 2)
Patients with primary PH also have some immunological disturbances, suggesting a possible role for inflammation in the pathophysiology of this disease. A subset of PH patients have been shown to have circulating autoantibodies, including antinuclear antibodies, as well as elevated circulating levels of the pro-infammatory cytokines, interleukins ‐1 and ‐6.
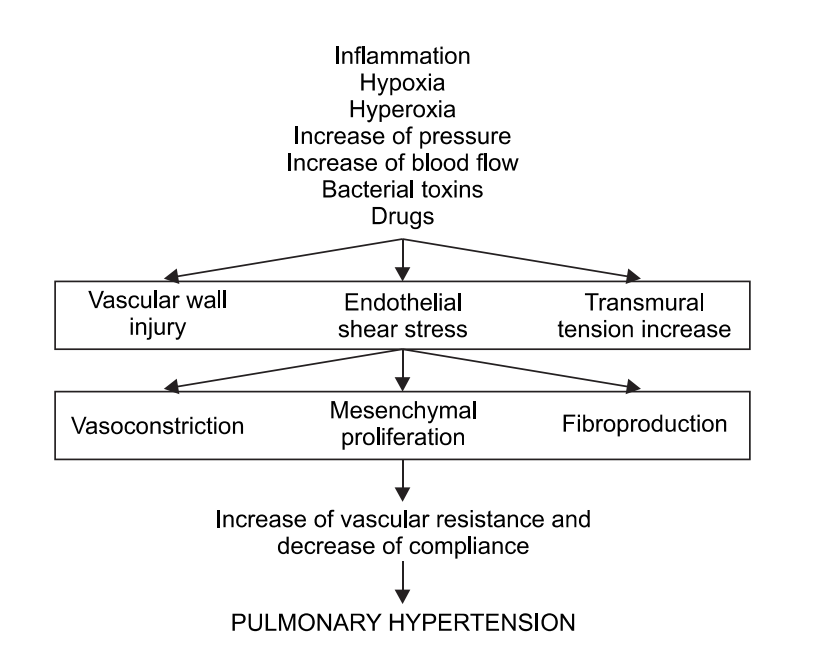
Figure 2
[5]
Pathogenesis of Pulmonary Hypertension
Pulmonary vasoconstriction is believed to be an early component of the pulmonary hypertensive process and may be related to abnormal function of potassium channels and endothelial dysfunction. Endothelial dysfunction leads to chronically impaired production of vasodilators such as nitric oxide and prostacyclin along with overexpression of vasoconstrictors such as endothelin.
PAH is characterized by three pathological characteristics viz. in situ thrombosis, smooth muscle hypertrophy and intimal and adventitial proliferation. Plexiform lesion which is frequently seen in PAH appears to be a dysfunctional response to vascular injury in these patients.
Key pathways involved in the pathogenesis of pulmonary arterial hypertension: a) endothelin (ET) pathway; b) nitric oxide pathway; and c) prostacyclin (PGI2) pathway. Increased number and density of ET (endothelin) receptors in the small pulmonary arteries of patients with idiopathic PAH is documented.
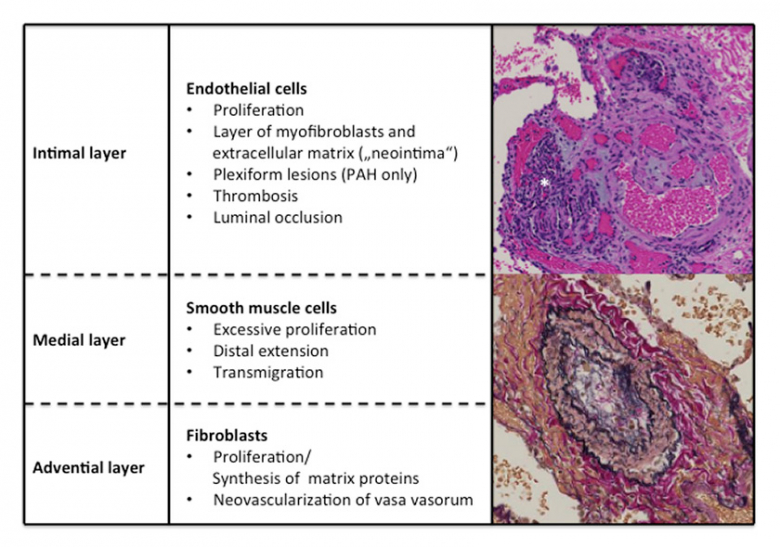
Figure 3: Pathological changes in pulmonary artery
|
Associated |
Likely |
Possible |
|
Aminorex |
Amphetamines |
Cocaine |
|
Fenfluramine |
Dasatanib |
Phenylpropanolamine |
|
Dexfenfluramine |
L-Trptophan |
St John`s wart |
|
Benfluorex |
Methamphetamines |
Interferon alpha and beta |
|
Selective serotonin reuptake inhibitors |
|
Mitomycin C |
|
Toxic rapeseed oil |
|
Cyclophosphamide |
Drugs and toxins causing PAH - Table 2
[7]
Genes associated with pulmonary arterial hypertension (as evident in Table 3)
Currently, a total of 14 PAH genes (ALK1, BMPR1B, BMPR2, BMP9, CAV1, EIF2AK4, ENG, KCNK3, KCNA5, KLF2, SMAD1, SMAD4, SMAD9, and TBX4) are known to be involved in the BMPR2 (Bone Morphogenetic Protein Receptor Type 2} signalling pathway. The following networks related to myofibroblast proliferation and vascular remodelling, whereas patients express strongly expressed proinflammatory genes.
|
Some Encoding Variation |
|
|
BMPR2 |
Bone morphogenetic protein receptor type 2. BMPR2, encoding bone the most commonly affected gene in both |
|
CAV 1 |
Encodes caveolin-1, relevant to caveolar structure as well as nitric oxide signalling |
|
ACVRL1 |
Activin A receptor like type 1 |
|
ENG |
Endoglin |
|
SMAD9 |
SMAD family member 9 |
Table 3
Diagnosing Pulmonary Hypertension : Doppler echocardiography suggest the presence of pulmonary hypertension, but right heart catherization remains the gold standard for diagnosis of PAH. Signs of pulmonary hypertension on CT scan of the chest are: Enlargement of the pulmonary trunk (measured at its bifurcation). It is, however, a poor predictor of pulmonary hypertension in patients with interstitial lung disease.
[8]
|
Class |
Drugs |
Feature |
|
|
Calcium channel blockers |
Amlodipine |
Only in Idiopathic Pulmonary Hypertension |
|
|
Endothelin Receptor Antagonists |
Bosentan, Sitaxantan, Ambrisentan |
Hepatic Toxicity |
|
|
Phosphodiesterase inhibitors |
Sildenafil, Verdanafil, Tadalafil |
No combination with NO donors |
|
|
Guanylate cyclase stimulators |
Riociguat |
No combination with NO donors |
|
|
Prostacyclin analogues |
Iloprost , Epoprostenol, Treprostinil |
IV, if SC is not tolerated |
|
|
Prostacyclin receptor agonists |
Selexipag |
Used orally |
|
Table 4 : Drugs in Pulmonary Hypertension ( Group 1) :
Nitric oxide: activates cGMP-dependent protein kinase, which inhibits Ca++ entry into the cell, reduces intracellular Ca++. In addition, intracellular cGMP activates myosin light chain phosphatase, reducing vascular tone.

Mode of Nitric Oxide action-Figure 4
NO reduces pulmonary resistance. Inhaled NO is the first vasodilator to produce selective pulmonary vasodilation. Usual dose of nitric oxide is 20 ppm. This produces maximal pulmonary vasodilatation in the vast majority of infants with pulmonary hypertension. Sudden discontinuation of iNO will cause rebound pulmonary hypertension that may be severe.
Inhaled O-nitrosoethanol gas (ENO) as a novel alternative means of providing nitric oxide bioactivity in the treatment of persistent pulmonary hypertension of new born.
[9]
Calcium channel blockers: are currently restricted to "responders" only. It is believed that no more than 5% of PAH patients will benefit from CCB long term. Ca blockers inhibit the calcium influx into vascular cells leading to relaxation of smooth muscle cells and vasodilatation. Vasoconstriction of small pulmonary arteries is recognized as a component of the pathogenesis of pulmonary arterial hypertension and treatment with calcium-channel blockers appears to be rational in this setting. PH will have a better prognosis when high dose of calcium channel blockers is used. Nifedipine and diltiazem are the most commonly used CCBs for PAH but may be prescribed in far higher doses (nifedipine up to 90mg daily, diltiazem up to 720mg daily) than for other uses such as angina. [10]
5 Phosphodiesterase inhibitors: Sildenafil is an inhibitor of cGMP specific phosphodiesterase type-5 (PDE5) in the smooth muscle of the pulmonary vasculature, where PDE5 is responsible for degradation of cGMP. Sildenafil, therefore, increases cGMP within pulmonary vascular smooth muscle cells resulting in relaxation. Oral sildenafil, however, caused pulmonary vasodilatation in patients with lung fibrosis and pulmonary hypertension, with an overall vasodilatory potency corresponding to that of intravenous prostacyclin. The recommended dose of Sildenafil is 20 mg three times a day (t.i.d.). Tadalafil is a selective long-acting PDE-5 inhibitor originally manufactured for the treatment of erectile dysfunction and was approved by the FDA in 2009 as a once-daily dose treatment for PAH. Tadalafil was approved as a once-daily dose of 40 mg taken orally with or without food. Vardenafil is effective and well tolerated in patients with PAH at a dose of 5 mg twice daily [11].
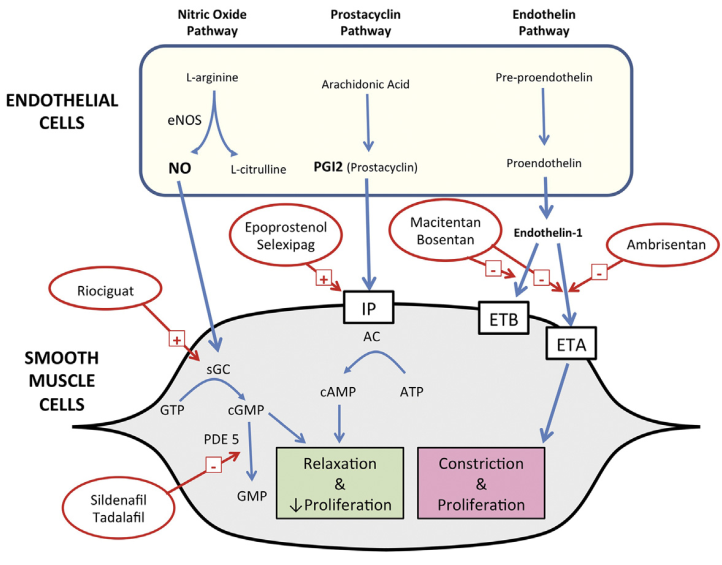
Actions of Riociguat, Prostacyclins and Endothelin receptor antagonists -Figure 5
Riociguat: The mechanism of action of riociguat is twofold. It binds sGC, resulting in vasodilation in the lungs from the stabilization of nitrous oxide (NO) binding to sGC, which sensitizes this pathway. In addition, it directly stimulates sGC independent of NO. Both mechanisms increase intracellular cGMP and relaxation of vascular tone.
Indicated for PAH, (Group 1), Oral riociguat administered t.i.d. was successfully titrated from a starting dose of 1 mg to a maximum target dose of 2.5 mg in a majority of patients.
[12]
CHEST-1 was a phase III study of Riociguat in 261 patients with inoperable ) chronic thromboembolic pulmonary hypertension or persistent/recurrent CTEPH after Pulmonary endarterectomy . After 16 weeks’ treatment with Riociguat (up to 2.5 mg tid), the primary end point of 6MWD increased by 46 m vs placebo (P < .001). CHEST 2 study has shown Long-term riociguat had a favourable benefit–risk profile and apparently showed sustained benefits in exercise and functional capacity for up to 1 year [13].
Prostaglandin analogues
Aerosolized iloprost: is a potent pulmonary vasodilator that is more effective in decreasing pulmonary artery pressure and increasing cardiac output than inhaled nitric oxide . Furthermore, inhalation of iloprost improves exercise capacity and oxygen uptake. Inhalation dose was 2 microg/kg b.w. and between 44 and 65 inhalations were performed in each patient starting within the first hour of life over a total of several days. Single inhalations lasted 5 min and were not repeated until 60 min had elapsed.
The 6-minute walk distance improved with treprostinil along with small, but statistically significant improvements in pulmonary arterial pressure and cardiac output.
Epoprostenol: directly vasodilates the pulmonary and systemic arterial vasculature, which reduces ventricular afterload, pulmonary vascular resistance and platelet aggregation, and increases cardiac output (CO). Intravenous epoprostenol has been shown to improve exercise capacity, hemodynamics, and survival in a short-term, randomized controlled trial in PPH. 2 ng/kg/min IV infusion pump over 24-48 hours; may initiate at lower dose if intolerant to starting dose. Titrate by 1-2 ng/kg/min q15min or longer, until desired effect or dose-limiting pharmacologic effects occur.
Selexipag: is Oral PGI2 receptor agonist. A Non-prostanoid pro-drug Its Metabolized rapidly to an acBve metabolite with high
affinity for the classical human “IP receptor” Metabolite has a long plasma half-life of 8hrs . Selexipag and its metabolite binds to other prostacyclin receptors with significant affinity. Selexipag may have a better side effects profile than other oral prostanoids. Dose of Selexipag varies from 200mcg bd to 800mcg bd
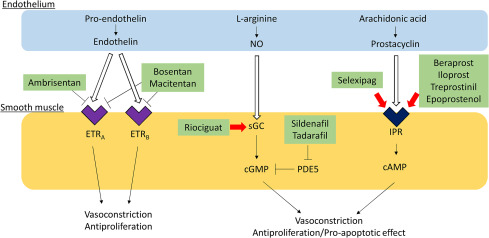
Action of Prostaglandin analogues -Figure 6
Beraprost sodium: is an orally active prostacyclin analog that has been reported in uncontrolled studies to improve hemodynamics in PAH. The maximal tolerated dose of beraprost sodium (median dose 120 microg four times a day) [14, 15. 16]
Endothelin receptor antagonists (ERAs)
Endothelin-1 (ET-1) is a 21 amino acid peptide that is produced by the vascular endothelium It is a very potent vasoconstrictor that binds to smooth muscle endothelin receptors, of which there are two subtypes: ETA and ETB receptors. Because of its powerful vasoconstrictor properties of Endothelin and its effects on intracellular calcium, ET-1 has been implicated in the pathogenesis of many vasoconstrictive diseases, including PAH.
Blocking endothelin receptors drug like ambrisentan counteracts many of the detrimental effects of endothelin, a vasoactive peptide that has a role in the pathogenesis of PAH.
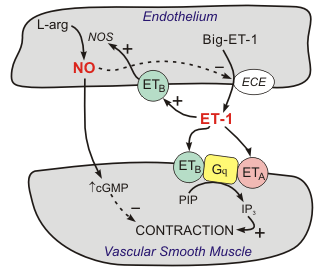
Action of Endothelin receptor antagonists -Figure 7
Endothelin is made in the layer of cells that line the heart and blood vessels. It causes the blood vessels to constrict (become narrower). In people with PH the body produces too much endothelin. This causes the blood vessels in the lungs to become narrow, increasing the blood pressure in the pulmonary arteries. ERAs reduce the amount of endothelin in the blood, therefore limiting the harm an excess of endothelin.
As the selective ET-A antagonist Darusentan reduced blood pressure who are difficult to treat and who are at increased vascular risk, e. g. patients with diabetes mellitus
|
Bosentan |
Sitaxantan |
Ambrisentan |
|
Etherocyclic sulphonamide |
Amidothiophene sulphonamide |
Diphenylpropanoic acid |
|
125 mg BD |
!00mg Qd |
5-10mg Qd |
|
No ETA selectivity |
ETA selectivity |
ETA selectivity |
Table 5: Dose of ERA
All three drugs are contraindicated in conjunction with cyclosporine A and glyburide [17]
Other potential therapeutic targets: There are a few clinical reports of stem cell therapy in patients with PAH. In a 12-week open-label controlled study, infusion of autologous endothelial progenitor cells (EPCs) improved 6MWD and haemodynamic variables in adult patients with severe PAH
Aromatase converts androgens to oestrogen and is evident in the pulmonary vasculature of PAH lungs. A small trial of Anastrozole, an aromatase inhibitor, in PAH has reported an increase in 6MWD (Six minute walking distance).
Administration of 3 months of Ranolazine therapy, accompanied by stable background pulmonary vasodilator therapy, was associated with improvements in RV structure and systolic function both at rest and during exercise. Ranolazine in PAH appears safe, without acute hemodynamic effects after a 500-mg dose.
Trimetazidine improved right ventricular function through indirect effect of increased glucose oxidation by blocking the Randle cycle. Trimetazidine can improve right ventricular function in pulmonary arterial hypertension patients.
Imatinib has been shown to suppress upregulation of PDGF and improve haemodynamic parameters in some patients with PAH. Nilotinib was more effective than imatinib in reducing right ventricular pressure.
Selective serotonin reuptake inhibitors use has been shown to be associated with reduced mortality in patients with PAH, although evidence in favour of their use is not strong
Terguride inhibited the proliferation of pulmonary artery smooth muscle cells and abolished 5-HT-induced pulmonary vasoconstriction in monocrotaline-induced pulmonary hypertension.
Sorafenib was well tolerated at 200 mg twice daily, and is currently under investigation in a phase I/II safety and tolerability study in patients with PAH already on existing therapy with a prostacyclin with or without sildenafil.
Sapropterin for 8 weeks in combination with sildenafil and/or an ERA resulted in improvements in 6MWD ( six minute walk distance) and was well tolerated.
Biological in Pulmonary arterial disease: Interleukin-1 (IL-1) and interleukin-6 (IL-6) are inflammatory cytokines and their levels correlate with survival in PAH. Anakinra is a recombinant form of the naturally occurring IL-1 receptor antagonist. Anakinra was well tolerated without any SAEs and it significantly reduced serum CRP.
TNFα inhibitor, Etanercept, was able to reverses disease progression in the /Hypoxia rat model of experimental PH
IL6 pathway is another potential target for PAH treatment because animal models have implicated a role for IL6 in PAH . Tocilizumab (8 mg/kg monthly) alters pulmonary vascular disease
Ubenimex is an oral, small-molecule inhibitor of leukotriene A4 hydrolase (LTA4H), an enzyme that plays a key role in the formation of inflammation-causing agents known as leukortriene B4 (LTB4).
There are many ongoing trials with above biologicals and the initial results have been satisfactory, where the duration of study extended till 24 weeks, where the outcome evaluated was change in ventilatory efficiency & biomarkers in multicentre trials.
[18]
RO kinase inhibitors: are a new class of drugs that may be be useful in the treatment of PAH. Fasudil showed efficacy in human studies in Japan and the USA. RO kinase activity has also been linked specifically to a number of known effectors of PAH, including ET-1, serotonin and endothelial NO synthase. long-term Fasudil administration in patients with severe PAH is required to evaluate efficacy and the safety. Treatment with Fasudil induced a marked improvement of medial wall thickening of pulmonary arteries partly due to its enhancing effect on Vascular Smooth Muscle Cell apoptosis. Hydroxyfasudil, an oral metabolite of fasudil, is a specific Rho-kinase inhibitor [19]

Table 6 : Algorithm for treating Pulmonary Arterial Hypertension [20]
V̇O2 max (also maximal oxygen consumption, maximal oxygen uptake or maximal aerobic capacity) is the maximum rate of oxygen consumption measured during incremental exercise; that is, exercise of increasing intensity. V̇Oi2 max is reduced in PAH. Functional class II patients were classified as intermediate/high risk.
Herbal drugs and nutraceuticals for PAH:
The active components in garlic are allicin, flavonoids and sulfur-containing proteins. The most active compound, Allicin, has been demonstrated to have anti-TNF-alpha properties, and therefore anti-inflammatory effects, in human studies
Quercetin is a flavonoid found in a wide range of plants and fruits with many biological activities such as antioxidant, anti-inflammatory, antitumor, antiproliferative, inducing apoptosis, antimetastatic, vasodilator and decreasing blood pressure. Also, it has been suggested that quercetin inhibits the progression of PH in some preclinical studies.
Radix Astragal a Chinese herb is used for the treatment of PAH, and its mode is multiple inhibition of the remodeling of intra-acinar pulmonary arteries and the hyperplasia of collagen and decrease in the content of endothelin-1( ET-1 ) and increase in the content of NO
Beet juice is commonly used as a supplement because of its high quantity of inorganic nitrate, a compound found naturally in vegetables and processed meats. Nitrate is believed to produce cardiovascular benefits,

Mode of action of Beet -Figure 8
Sulforaphane (1-Isothiocyanato-4-(methanesulfinyl)butane) is an active molecule found particularly in broccoli and cauliflower extracts that acts as an inducer of NAD(P)H quinone oxidoreductase 1 and quinone reductase activity The protective effects of these properties have been reported to occur in RV hypertrophy in PAH
Ginkgo biloba leaf reduce atherosclerotic nanoplaque formation and size, suppress atherosclerotic lesion development. Release of NO and cytokines, such as prostacyclines are stimulated.
The major components of Astragalus membranaceus are polysaccharides, flavonoids, and saponins.Contemporary use of Astragalus membranaceus mainly focuses on its immunomodulating, anti-oxidant, and anti-inflammatory effects.
Ligusticum wallichii Rhizome ie Ligustrazine, known as tetramethylpyazine, is a Ca2+channel antagonist which can dilate the blood vessels during hypoxia by blocking Ca2+ influx. ligustrazine can significantly reduce mean pulmonary arterial pressure. and the plasma endothelin-1 (ET-1) levels in acute hypoxia-induced pulmonary hypertension.
Tetrandrine (TET) is a bisbenzylisoquinoline alkaloid extracted from the root of Stephania tetrandra. TET acts as an antagonist of some vasoconstriction factors such as platelet-activating factor, angiotensin II, and prostaglandin F, which play a significant role in the occurrence and development of PAH.
Uncaria rhynchophylla The signalling pathways involved for exerting its vasodilatory effects includenitric oxide/soluble guanylyl cylcase/cyclic guanosine monophosphate (NO/sGC/cGMP) and PGI2 (endothelium-derived relaxing factors) [21]
Pulmonary endarterectomy (PEA) surgery involves clearing all obstructive thromboembolic material from the pulmonary arteries, including the intima and superficial media. Effective PEA intervention can result in near normalisation of pulmonary haemodynamics, with significant and immediate reductions in PVR and mean pulmonary arterial pressure (mPAP), together with an increase in cardiac index . PEA is the gold standard treatment for ( CTEPH) chronic thromboembolic pulmonary hypertension as it is potentially curative.
Transplantation remains the therapeutic option for selected patients with advanced pulmonary arterial hypertension (PAH) who continue to deteriorate despite optimal pulmonary vasodilator therapy – including combination therapy.
Guidelines for Lung Transplant include rapidly progressive disease , low or declining 6 minute walk , Cardiac index of less than 2 litre / min / m2 , Right arterial pressure > 15mmHg , NYHA functional class III or IV , irrespective of ongoing therapy. Poor outcomes in PAH such as hyponatraemia and hyperbilirubinemia, patients not responding to any medical therapy.
Double-lung transplantation is the recommended operation for idiopathic PAH. Lung transplantation provides an opportunity for patients to extend their lives as well as improve quality of life. According to International Society of Heart Lung Transplantation (ISHLT) registry data, overall median survival for all lung transplant recipients transplanted was 5.3 years, and among those who survive at least 1 year, median survival was 7.5 years. [22, 23 ]

Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org