Pediatrics and Child Health Issues
OPEN ACCESS | Volume 6 - Issue 1 - 2026
ISSN No: 2836-2802 | Journal DOI: 10.61148/2836-2802/JPCHI

Shamma AlZaabi1, Abdul-Kader Souid2*
Department of Pediatrics, College of Medicine and Health Sciences - UAE University, Alain City, United Arab Emirates
*Corresponding author: Abdul-Kader Souid, Department of Pediatrics, College of Medicine and Health Sciences - UAE University, Alain City, United Arab Emirates
Received: March 14, 2021
Accepted: March 25, 2021
Published: April 19, 2021
Citation: Shamma AlZaabi, Abdul-Kader Souid, “A Brief Guide on Pediatric Hematology”, J Pediatrics and Child Health Issues,2(1); DOI: http;//doi.org/03.2021/1.1014.
Copyright: © 2021 Abdul-Kader Souid. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly Cited.
,
1. Interpretation of the Pediatric Complete Blood Count
Reference (‘normal’) values of the complete blood count (CBC) vary with age and gender. Hematocrit, mean cell hemoglobin (MCH) and mean cell hemoglobin concentration (MCHC) are calculated quantities and have limited clinical use. Moreover, the white blood cell (WBC) count, the sum of the more important differential counts, has also limited clinical use. Percentage counts are not needed [1].
Proper clinical interpretation of the neutrophil, lymphocyte, monocyte, eosinophil, and basophil counts is essential. For example, a consistent neutrophil or lymphocyte count of <1.0 x109/L always signifies neutropenia or lymphopenia (T cell deficiency), respectively. Moreover, a normal differential count does not contain nucleated red cells, myelocyte or metamyelocyte (‘left shift’). A normal platelet count is 150 to 400 x109/L; values <150 x109/L are thrombocytopenia and >400 x109/L are thrombocytosis (most commonly reactive).
Hemoglobin concentration. Normal hemoglobin concentration varies with age. Higher values occur in the newborn and in the adolescent male (Table 1). The low fetal arterial oxygen tension (PO2) of ~30 mm Hg stimulates erythropoietin secretion, leading to secondary polycythemia. After birth, the rise in arterial PO2 to ~90 mm Hg suppresses erythropoietin secretion, leading to a gradual decline in the red cell production that nadirs at ~2 months (physiologic anemia). This effect is steeper and occurs earlier in preterm neonates (with a typical hemoglobin concentration of 80 to 90 g/L by 1 to 2 months). Subsequently, a steady rise in hemoglobin concentration reaches maximum in post-pubertal males due to the effects of testosterone. The blood volume of a neonate increases by allowing emptying of placental vessels before cord clamping; a maximum delay in cord clamping of 60 seconds is recommended. Hemoglobin concentration is also influenced by the blood sampling technique; heel stick samples are less reliable than venous samples.
|
Table 1. Lower limits (-2 SD) of hemoglobin concentration (g/L) |
|
|
Birth |
145 |
|
2 to 6 mo |
90 (physiologic anemia) |
|
4 to 12 y (pre-pubertal) |
115 |
|
12-18 y (pubertal and post-pubertal males) |
140 |
|
12-18 y (pubertal and post-pubertal females) |
123 |
Red blood cells (RBC). The normal RBC count ranges from 3.5 to 6.5 x1012/L. About 1% to 2% of the circulating red cells are reticulocytes (immature non-nucleated red cells containing ribonucleic acid). Thus, the normal reticulocyte count ranges from 35 to 130 x109/L; values <35 x109/L are reticulocytopenia and >130 x109/L reticulocytosis (Table 2). The nadir of reticulocyte count occurs between one week and 2 months, which results in physiologic anemia; a high reticulocyte count during this period signifies hemolysis. Nucleated red cells are seen in the first week of life; their presence after one week signifies prenatal/ perinatal hypoxia, hemorrhage or hemolysis. After the newborn period, circulating nucleated red cells indicate increased demand for production (e.g., hemorrhage or hemolysis) or bone marrow destruction (infiltration).
|
Table 2. Normal reticulocyte counts |
||
|
Birth |
100 to 500 x109/L |
3% to 7% |
|
1 week to 2 mo |
10 to 50 x109/L |
0.1% to 1% |
|
>2 mo |
50 to 100 x109/L |
1% to 2% |
Mean cell volume (MCV). Red cell volume is expressed in femtoliter (fL = 10-15 L). Its value is determined using a calibrated electrical impedance (mean height of the voltage pulses generated during red cell counting). The red cells are macrocytic at birth, with MCV of 98 fL to 116 fL. MCV <94 fL at birth (cord blood sample) is microcytosis and signifies a-thalassemia trait (since b-thalassemia and iron deficiency anemia do not present at birth), Table 3. After one month, MCV >98 fL indicates macrocytosis (e.g., B12 deficiency, which could be masked by a coexisting thalassemia trait, especially in a population with high prevalence of a-thalassemia trait). At one month of age, the lowest normal MCV is 70 fL. Thereafter, the lowest normal is “70 fL + y of age until 10 y”. Thus, MCV <70 fL is abnormal at any age.
|
Table 3. Normal MCV for age |
|
|
Birth (cord blood) |
98 to 116 fL |
|
1 mo to 10 y |
“70 fL + y of age” to 90 fL |
|
>10 y |
80 to 98 fL |
Relative distribution width (RDW = ‘standard deviation ÷ MCV’ [unitless, expressed as percentage) is a measure of the variation of red cell volume (relative standard deviation, coefficient of variation, degree of spread/ dispersion around the mean, anisocytosis). Normal RDW is <15% (or <0.15). Iron deficiency anemia is usually associated with increased RDW, signifying a mixture of red cells of various sizes. RDW is the first parameter to become abnormal in iron deficiency anemia and the last to correct. A simplified report of the CBC is shown in Table 4.
|
Table 4. Suggested CBC format |
|
|
Variables |
Units |
|
Hemoglobin |
g/L |
|
Red cells |
x1012/L |
|
Reticulocytes |
x109/L |
|
Standard deviation (SD) of red cell volume |
fL |
|
MCV |
fL |
|
RDW (SD of red cell volume ÷ MCV) |
Unitless |
|
Platelets |
x109/L |
|
Neutrophils |
x109/L |
|
Lymphocytes |
x109/L |
|
Monocytes |
x109/L |
|
Eosinophils |
x109/L |
|
Basophils |
x109/L |
II. Microcytic, Macrocytosis and Normocytic Anemias
Microcytosis (small red cells) signifies a decreased hemoglobin synthesis, usually due to iron deficiency (e.g., nutritional, milk-induced colitis, or hemorrhage), thalassemia trait (decreased a or b chain synthesis), chronic inflammation (blocks iron delivery to erythroid precursors), or [rarely] ineffective protoporphyrin
synthesis (e.g., hereditary sideroblastic anemia). Macrocytosis (large red cells), on the other hand, signifies impaired red cell production (e.g., adverse effect of a medication, B12 deficiency, or bone marrow failure syndrome). Normocytic (normal red cell size) hemolytic anemia is associated with jaundice, indirect hyperbilirubinemia, reticulocytosis, and splenomegaly. Examples include hemoglobinopathy (e.g., sickle cell disease), enzymopathy (e.g., glucose-6-phosphate dehydrogenase [G6PD] deficiency), cytoskeletal (membrane) abnormality (e.g., hereditary spherocytosis), and autoimmune hemolytic anemia.
III. Disorders Related to Iron
Iron bioavailability requires: (1) Ferritin, (2) Transferrin, (3) Transferrin receptor, and (4) Iron-responsive element (IRE) binding protein (IRE-BP, an mRNA-binding protein that senses cellular iron). Ferritin sequesters cellular iron for prompt mobilization when needed. Transferrin carries iron in the plasma. Transferrin receptors bind and internalize the transferrin-iron complex. IRE-BP coordinates the production of ferritin and transferrin receptors. If cellular iron is low, IRE-BP binds to IRE in the 5’ untranslated regions of mRNA, stimulating the translation of transferrin receptor and inhibiting the translation of ferritin; thus, promoting iron uptake and decreasing iron storage. The opposite occur in iron overload.
The “divalent metal transport protein-1” (DMT-1) is found on the apical membrane of enterocytes (mainly in the duodenum and upper jejunum); it mediates cellular Fe2+ uptake. Iron then exists the cell via the transmembrane exporter ferroportin. Ferroportin is inhibited by hepcidin [2]. Of note, celiac disease should be considered in the diagnostic evaluation of children with iron deficiency [3-4]. Their oral iron is unlikely to be absorbed until the intestines recover (with the elimination of gluten). For these children, parenteral iron is recommended [5-6].

The gastric acid, ascorbic acid, citric acid (juices), lactose (sugars) reduce Fe+3 to Fe+2, thus, increasing the bioavailability of dietary non-heme iron. Ascorbic acid also prevents binding of Fe+3 to plant phytates found in grains, such as oat, and rye, and polyphenols (e.g., tannins found in green tea, which binds iron and lowers its absorption).
Hepcidin, a critical regulator of iron bioavailability, is a hepatic hormone stimulated by iron and inflammation, and suppressed by iron deficiency, erythropoietin, hypoxia, and ineffective erythropoiesis [2]. Hepcidin inhibits iron absorption by binding to ferroportin and causing its internalization and degradation. Hepcidin also reduces iron efflux from macrophages and hepatocytes. Moreover, cytokines (e.g., lipopolysaccharide and interleukin-6) induce the release of hepatic hepcidin, resulting in iron sequestration in macrophages and ‘anemia of inflammation’. Low hepcidin, on the other hand, leads to increased iron absorption and export.
Absorbed Fe+2 is either stored as ferritin or exists the enterocyte through the basolateral membrane into the blood capillary; this transport is mediated by ferroportin (depicted in the shown schema). Ferroportin is present on the basolateral membrane and the membranes of macrophages, hepatocytes, and placenta. In the blood, transferrin binds exported iron and delivers it to the bone marrow and liver via the cell surface transferrin receptors.
Physiologic responses to iron deficiency include: (1) Low serum ferritin (levels <12 µg/L indicate absent iron stores), (2) Low serum iron (<40 μg/dL), (3) Low transferrin saturation (the ratio of plasma iron to total iron-binding capacity [TIBC], which reflects iron supply to tissue; normal = 35% ± 15%; <16% is a criterion for iron-deficiency), (4) High soluble transferrin receptors (sTfR, normal = 5.5 mg/L), (5) High zinc protoporphyrin (zinc replaces iron on protoporphyrin; high levels are seen with inflammation, lead poisoning and sideroblastic anemia; normal value is 30 μg/dL (0.53 μmol/L), which increases to 100 μg/dL [1.78 μmol/L] in iron deficiency), (6) Anemia (normocytic → microcytic anemia), (7) High RDW, and (8) Reticulocytopenia [7-8]. It is important to note that serum ferritin, fasting serum iron, transferrin, and transferrin saturation are unreliable determinants of iron overload.
Inflammation and damages to ferritin-rich tissues (e.g., liver) elevate serum ferritin independent of iron stores. Serum ferritin is increased in inflammation; sTfR is not affected by inflammation; thus, sTfR is used to differentiate anemia of inflammation from iron deficiency.
MRI-based Ferriscan is a reliable indicator of liver iron. Children with thalassemia major require iron chelation when serum ferritin is >1,000 ng/mL. MRI-based Ferriscan scan for liver iron quantitation is then requested at annual intervals to guide iron chelation. If the estimated liver iron is >15 to 25 mg Fe/g dry weight liver, MRI-based Ferriscan quantitation of cardiac iron should also be done. Chelation with one or two drugs should be utilized to maintain liver iron between 3 and 7 mg Fe/g dry weight liver and normal cardiac iron. Of note, liver iron (measured in a biopsy sample) is limited by the variable iron content throughout the liver (e.g., missing foci of hepatic fibrosis).
Transfusional iron (expressed in ‘mg iron per kg’) can be estimated as: ‘Total volume of transfused red cells (in mL) x 0.6 (the average hematocrit in red cell bags) x 0.8 to 1.0 mg (the amount of iron per mL of transfused red cells; as 0.8 to 1.0 mg iron is acquired for every mL of red cells transfused) ÷ patient weight (kg)’. Liver iron (expressed in ‘mg iron per g dry liver weight’) is estimated as: ‘Transfusional iron (mg/kg) ÷ 10.6’. Liver iron content of 15 to 20 mg/gram dry weight is associated with liver fibrosis. Chelation is recommended when liver iron is >7 mg/g dry weight liver.
Children with ‘ineffective erythropoiesis’, such as thalassemia intermedia should have their first liver Ferriscan at 10 y of age and
every year thereafter depending on the results. In the absence of any transfusion, their mean annual increase of hepatic iron typically is 0.38 ± 0.49 mg Fe/g dry weight liver, which is mediated by suppressed hepcidin, causing increased iron absorption. At this rate, iron accumulation leads to serious complications in young adults, such as hepatic fibrosis, cardiac dysfunction, renal tubular dysfunction, hypogonadism, diabetes, hypoparathyroidism, and infertility.
Haptoglobin (the primary hemoglobin-binding protein in plasma) and hemopexin (promotes detoxification of heme when haptoglobin is depleted) are responsible for intravascular iron salvages. Haptoglobin and hemopexin are acute phase reactants; their levels are increased in infection/ inflammation. Both haptoglobin and hemopexin prevent free hemoglobin from being filtered through the kidney. Haptoglobin is the major hemoglobin-binding protein during intravascular hemolysis. Free hemoglobin binds to haptoglobin; while free heme binds to hemopexin. The heme-hemopexin complex is metabolized in the liver. The hemoglobin-haptoglobin complex (>150 kD) is internalized by CD163 (the monocyte-macrophage surface receptor) in ‘red pulp’ macrophages of the spleen and in Kupffer cells of the liver. The CD163 gene is induced by glucocorticoids, increasing the clearance of hemoglobin-haptoglobin.
About 70% of the body iron is in heme (hemoglobin and myoglobin), 29% in ferritin or hemosiderin (storage forms of iron), and <1% in heme-containing enzymes (cytochromes, catalase, peroxidase) or in the plasma as transferrin. Daily iron loss is ~2 mg; mostly from sloughing in the gut, skin, and renal tubules; 6 µg iron per kg per day (<0.5 mg per day) is lost during normal menstruation.
An average diet contains two forms of iron: (1) Heme (10%, mainly Fe2+) and (2) Non-heme (90%, mainly Fe+3). Only 1-2 mg (10% of dietary iron) is absorbed daily. About 25% of heme iron (meat, poultry and fish) is absorbed, while only 5% non-heme iron (cereals, beans, and vegetables) is absorbed.
IV. Iron deficiency anemia (IDA)
Iron deficiency anemia (IDA) is common in early childhood. It is characterized by decreased hemoglobin, low MCV (microcytosis due to decreased hemoglobin synthesis), high RDW (heralds the production of smaller red cells), and reticulocytopenia (iron-limiting erythropoiesis). Children between 6 mo and 3 y are especially susceptible due to inadequate dietary iron for their rapid growth. In the first 4-6 mo, most full-term infants have adequate iron transferred through the placenta during the last trimester of pregnancy. Thus, premature infants frequently develop iron deficiency. In infants, cow’s milk induces colitis (gastrointestinal bleeding); thus, whole milk should not be introduced in the first year. The most conclusive evidence of iron deficiency anemia is iron-responsive anemia (iron-rich food [preferred as other micronutrient deficiencies are likely to coexist] with or without iron supplementation [depending on the severity]). Optimal response is obtained with 2 to 6 mg/kg/day of elemental iron; ferrous sulfate is the preferred form. The reticulocyte count increases in one week, followed by a gradual correction over 4 weeks. Treatment should continue for 3 mo [9-10].
Recommendations to prevent iron deficiency include iron supplement (1 to 2 mg/kg/day) to exclusive breast-fed infants after 4 mo; use of iron-fortified formulas (12 mg/L iron as ferrous sulfate) and cereals; iron supplement (2 to 3 mg/kg/day) for preterm infants after first mo; and delay cow’s milk until after first year [9-10].
Iron deficiency may result in impaired growth, poor school achievement, delayed motor and cognitive development, impaired immune responses, hyperactivity, and socio–emotional problems. Lower scores in the Bayley Scales of Infant Development have been demonstrated at 9 to 12 mo between non-anemic, iron deficient, and iron-sufficient infants. Dietary supplements from early age is a public health solution to this common problem [11-12].
V. Thalassemia
(Greek: thalatta or thalassa = sea’ –aima = blood; originating around the Mediterranean and Black Seas, hence the origin of the name, “Mediterranean Anemia”)
These inherited disorders are characterized by decreased synthesis of the a- or b-chain. Synthesis of the a-chain peaks at 3 mo of fetal age and b-chain peaks at 3 mo after birth. Thus, a-thalassemia manifests at birth and b-thalassemia after 3 mo of life. Single gene copies of the b (hemoglobin-beta locus; HBB, MIM#141900), d (hemoglobin-delta locus; HBD, MIM#142000), g (hemoglobin, gamma A; HBG1, MIM#142200 and hemoglobin, gamma G; HBG2, MIM#142250), and epsilon (e) are mapped to each allele of chromosome 11 (as shown in the schema).
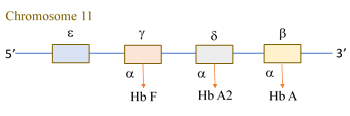
Two copies of the a gene (hemoglobin-alpha locus 1; HBA1, MIM#141800 and hemoglobin-alpha locus 2; HBA2, MIM#141850) are mapped to each allele of chromosome 16. Thus, a normal individual has four a genes. The four chains (a, b, g, and d) form the three normal hemoglobin variants (Table 5): (1) Hb A (93% to 97%, two a plus two b), (2) Hb A2 (two a plus two d), and Hb F (two a plus two g). Since Hb A2 and F do not contain b chain, b-thalassemia trait is associated with higher percentages of A2 and F. In a-thalassemia, the percentages of Hb A2 and F remain normal. Thus, accurate determination of A2 is crucial, as a slight (≥0.5%) elevation of A2 signifies b-thalassemia trait. Children with b-thalassemia major (homozygous b-thalassemia, two defective b genes) have severe microcytic anemia and depend on blood transfusion. The variant either abolish (b0) or reduce (b+) b-chain synthesis.
|
Table 5. Normal values of hemoglobin A2 and F variant as function of age. |
|
|
A2 |
1-30 days = 0.0-2.1%; 1-2 mo = 0.0-2.6%; 3-5 mo = 1.3-3.1%; ≥6 mo = 2.0-3.3%. |
|
F |
1-30 days = 22.8-92.0%; 1-2 mo = 7.6-89.8%; 3-5 mo = 1.6-42.2%; 6-8 mo = 0.0-16.7%; 9-12 mo = 0.0-10.5%; 13-17 mo = 0.0-7.9%; 18-23 mo = 0.0-6.3%; ≥24 mo = 0.0-0.9%. |

There are four a-thalassemia syndromes (see schema), as follows: (1) Silent carrier involves one deleted gene. These individuals have low-normal MCV or mild microcytosis (75 fL to 80 fL). (2) a-Thalassemia trait involves two deleted genes. These individuals have mild-to-moderate microcytosis (MCV <75 fL). There are two genetic forms of a-thalassemia trait. The cis-form involves two deleted genes, ‘both on one allele’; this serious form is common in the Mediterranean population. The trans-form involves two deleted genes, ‘one on each allele’; this benign form is common in the Arabian Peninsula. (3) Hemoglobin H disease involves three deleted genes (one parent has cis-form a-thalassemia trait [heterozygous a0] and one has silent carrier). The unpaired b-chains form tetramers (b4), termed "hemoglobin H" (not to be confused with hemoglobin H disease). The b4 aggregates are more soluble than the a4 aggregates. Thus, patients with hemoglobin H disease usually have only moderate-to-severe hemolytic anemia. (4) Hydrous fetalis involves four deleted genes (both parents have ‘heterozygous a0’). The unpaired g-chains (g4, ‘Bart’s hemoglobin, a fast migrating variant’) have high affinity to oxygen (bind oxygen tightly and release almost none to the fetal tissue), leading to hypoxia and hydrops (edema due to heart failure).
Bart’s hemoglobin is elevated in all forms of a-thalassemia; it can be used to screen for neonatal a-thalassemia. Bart’s Hemoglobin constitutes 1% to 2% in silent carrier, 2% to 10% in trait, 20% to 30% in hemoglobin H disease, and >80% in hydrops fetalis. As noted above, newborns with a-thalassemia trait have MCV <94 fL at birth, which also serves the diagnosis of a-thalassemia trait. Stem cell transplantation is curative for thalassemia major.
VI. Chronic transfusion
Complications of chronic transfusions include iron overload, alloimmunization, and viral infections. Accumulation of iron results in diabetes and hypopituitarism (hypogonadotropic hypogonadism, impaired fertility, osteopenia, and osteoporosis). Cardiomyopathy and arrhythmias are the leading causes to death. The same risk for hypogonadism exists with stem cell transplantation and is caused by previous iron overload and the conditioning regimen. One-third of females and two-thirds of males with thalassemia major fail to enter puberty after myeloablative treatment. Patients who are compliant with chelation have 50% chance of being alive at 30 y, whereas for noncompliant patients, this rate is 10%. Transmission of viral infections, especially HIV, hepatitis B, and hepatitis C remains an important complication.
Iron chelation is recommended for children >2 y after >10 transfusions, serum ferritin >1,000 µg/L (2,247 pmol/L), or liver iron ≥3 mg/g dry weight liver. The goals of chelation are liver iron 3 to 7 mg/g dry weight liver, ferritin 1,000 ng/mL (2,247 pmol/L), cardiac T2* >20 ms, and avoidance of organ dysfunction caused by iron overload. With aggressive chelation, iron-induced cardiotoxicity can be reversed and some endocrine complications may be improved. It is necessary to monitor for the adverse effects of chelation therapy with monthly liver and kidney function tests as well as annual audiologic and vision screening.
VII. Neonatal hematology
Hemoglobin F does not bind to 2,3-bisphosphoglycerate (2,3-BPG), accounting for its high oxygen affinity compared to hemoglobin A. This physical property allows hemoglobin F to bind more oxygen. Hemoglobin F is also more easily oxidized to methemoglobin (unable to bind oxygen as the iron in the heme is ferric [Fe3+] rather than ferrous [Fe2+]). Cells rich in hemoglobin F are larger and more resistant to osmotic stress than adult red cells. Thus, the diagnosis of hereditary spherocytosis utilizing osmotic fragility test should be delayed until after 6 mo.
Anemia of prematurity occurs in infants ≤32 weeks’ gestation. The hemoglobin reaches nadir at 4 to 8 weeks, as compared to the physiologic anemia of full-term newborns at 8 to 12 weeks. Its etiology includes low erythropoietin secretion and frequent phlebotomy. Prevention includes: (1) delayed cord clamping (30 to 60 sec), (2) reducing phlebotomy, (3) providing iron, and (4) increasing protein intake (3 to 4 g/kg/day).
Alloimmune hemolytic disease of the newborn (erythroblastosis fetalis) is a severe hemolysis caused by maternal anti-D (in Rh D negative mother) against D antigen (in Rh D positive fetus). Maternal anti-Kell is the second most common. Severity of the anti-Kell disease may progress quickly due to antibody-mediated suppression to erythropoiesis. Maternal antibodies against Duffy and the c and E antigens in the Rh group can also cause severe hemolysis. These infants are at risk for hyperbilirubinemia (kernicterus). Infants of these mothers may require an exchange transfusion if the bilirubin level is concerning. If the disorder is identified in utero, intrauterine transfusion may be utilized. IgG is not beneficial. Patients must be followed closely because anemia may be lasting. The primary approach to RhD incompatibility is
prevention; an RhD-negative mothers should receive one dose of anti-RhD at 28 weeks’ gestation and again postnatally. Hemolytic disease of the newborn caused by ABO incompatibility occurs exclusively in association with type ‘O’ mothers and type ‘A’ or ‘B’ mother; it is usually mild.
Hereditary spherocytosis {spherocytosis type 1; SPH1 (MIM#182900, AD); ANK1 (MIM#612641, ankyrin 1); SPH4 (MIM#612653, AD); SLC4A1 (MIM#109270, solute carrier family 4, anion exchanger, member 1)} is a heterogeneous defect in the red cell cytoskeleton, most commonly due to abnormal spectrin. It is sporadic in 10% of the cases. The defect results in membrane (surface) loss, causing sphere-shaped cells, increased fragility and premature trapping in the spleen. The presence of spherocytes on the blood smear establishes the diagnosis. Hemolysis may begin in the first 24 h of life, causing neonatal jaundice. Hypoplastic episodes (severe anemia and reticulocytopenia) due to a viral infection (e.g., B19 parvovirus) are common. Blood transfusion may be necessary during acute hemolytic or hypoplastic episodes. Splenectomy is necessary for persistent hemoglobin <100 g/L and reticulocyte count >10%. In neonates, eosin-5’-maleimide (EMA) is more reliable than peripheral smear (as neonatal erythrocyte morphology has significant variation) or osmotic fragility. Eosin-5’-maleimide binding is influenced by the MCV; thus, neonatal references (with a typical MCV of 105 fL) are needed to interpret the results. In a patient with a classic presentation and typical spherocytes on peripheral smear, particularly with a positive family history, no further diagnostic testing is required. Otherwise, a combination of eosin-5’-maleimide-binding and acidified glycerol reliably identifies hereditary spherocytosis, particularly in mild disease.
Hereditary elliptocytosis (HE) describes peculiar red cells with micropoikilocytes, elliptocytes, budding, and fragmentation. It is caused by pathogenic variants in the red cell membrane cytoskeletal proteins (e.g., α-spectrin, β-spectrin, protein 4.1R, glycophorin C). Its four types are: (1) Common HE; (2) Hemolytic HE; (3) Hereditary poikilocytosis/pyknocytosis (HPP); and (4) Spherocytic elliptocytosis (SE). Common HE (autosomal dominant) shows elliptocytes on blood smear in asymptomatic child (a mild hemolysis may occur with illnesses). Affected neonates may have severe hemolysis. Splenectomy may be required in HPP to improve the anemia; the MCV is typically very low (30 to 50 fL). SE is characterized by elliptocytes and microspherocytes (without poikilocytosis or fragments).
Neonatal alloimmune thrombocytopenia results from maternal antibodies against human platelet antigen (HPA, most commonly HPA-1a followed by HPA-5b and HPA-15b) that is present in the fetus and not in the mother. It can occur in the first pregnancy. The thrombocytopenia is often severe and may result in intracranial bleeding. Maternal platelet apheresis is effective. Other causes include maternal immune thrombocytopenia (ITP), asphyxia, pregnancy-induced hypertension, intrauterine growth retardation, viral infection (cytomegalovirus), sepsis, and trisomy 21.
About 10% of infants of mothers with ITP develop thrombocytopenia, which may occur after the mother’s platelet count is normalized. The clinical presentation is less severe than in ‘neonatal alloimmune thrombocytopenia’. The risk of intracranial hemorrhage is low and is not influenced by the mode of delivery. Platelet counts need to be checked from a cord blood sample and repeated every 2-3 days for the first week. Platelet counts usually reach a nadir in 2 to 5 days and recover by day 7; some infants remain thrombocytopenic for 3 mo. Cranial ultrasonography should be performed to assess intracranial hemorrhage. Infants with a platelet count of <50 ×106/L or with a clinical bleeding should be treated with intravenous immunoglobulin (1 g/kg/day for 2 days) plus corticosteroids. Platelet transfusion may be used for severe cases.
Newborns whose mothers have systemic lupus erythematosus (SLE) frequently develop skin lesions (erythematous, annular) and complete atrioventricular block. Mothers with Ro/SSA and La/SSB autoantibodies are more likely to have a newborn with lupus syndrome. The resulting anemia and thrombocytopenia respond to transfusion, corticosteroids and intravenous immunoglobulin. The neutropenia resolves by 6 mo [?].
Trisomy 21 is frequently associated with lymphopenia, abnormal thymus development and function, frequent respiratory infections, autoimmunity, and hematologic malignancy. The newborn screening for severe combined immune deficiency (SCID) is through identification of T-cell receptor excision circles (TREC).
Severe congenital neutropenia is a heterogeneous disorder associated with neutrophil counts <0.2 x109/L from birth, myeloid arrest at the promyelocyte/myelocyte stage and recurrent infections. These infants have increased risk of myelodysplasia and acute myelogenous leukemia (AML). The use of granulocyte colony-stimulating factor (G-CSF) increases the risk of AML. In one study, there was a 5.7 higher risk of myelodysplasia/AML in children who received G-CSF at 15 μg/kg per day, compared with those who received no or lower G-CSF dose. The estimated hazard risk is 2% per year. Thus, G-CSF is given only for a specific infection. ELANE (elastase, neutrophil-expressed, MIM#130130) variants are seen in both severe congenital neutropenia (MIM#202700, autosomal dominant) and cyclic neutropenia (MIM#162800, autosomal dominant). Shwachman syndrome (gene: ribosome maturation factor, SBDS, MIM#607444; phenotype: MIM#260400, autosomal recessive) is associated with pancreatic malabsorption, bell-shaped chest, metaphyseal dysplasia, and waxing and waning neutropenia.
Neonatal alloimmune neutropenia occurs with maternal sensitization to fetal neutrophils bearing paternal antigens; more common in multiparous mothers. It may last up to 6 mo. The bone marrow shows myeloid hyperplasia. The antibody is identified in the serum of the mother and the infant, and is against the HNA-1 neutrophil FCγ receptor IIIb.
Benign (ethnic, familial) neutropenia is mild and associated with African, Yemenite, West Indian, and Arab ancestries. It is linked to variants in ACKR1 (atypical chemokine receptor 1; MIM#613665); previously termed: Duffy antigen receptor for chemokines (DARC).














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org