Pediatrics and Child Health Issues
OPEN ACCESS | Volume 4 - Issue 1 - 2025
ISSN No: 2836-2802 | Journal DOI: 10.61148/2836-2802/JPCHI

I.M.Rogers
Anaesthesiology, 46 Whit burn Road, Cleadon, Tyne and Wear SR67QS, Sunderland, UK
*Corresponding author: I.M.Rogers, Anaesthesiology, 46 Whit burn Road, Cleadon, Tyne and Wear SR67QS, Sunderland, UK.
Received date: December 03, 2020
Accepted date: December 11, 2020;
Published date: December 14, 2020
Citation: I.M.Rogers. “The Cause of Pyloric Stenosis of Infancy-Perspectives in Time.’’. J of Pediatrics And Child Health Issues, 1(1); DOI: http;//doi.org/03.2020/1.1002.
Copyright: © 2020 I.M.Rogers. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
A review is given of how knowledge about pyloric stenosis of infancy (PS) has evolved from its earliest discovery.
The historical evidence that underpins the development of the Primary Hyperacidity Theory is discussed in detail. Viewed from the perspective of this theory, all the previously unexplained features of the condition become understandable. The pyloric tumour develops because of exposure to a temporary hyperacidity which causes repeated sphincter contraction at a time in normal development when plasma gastrin levels are high. The associated trophic effect encourages extreme sphincter work hypertrophy.
The contemporary alternative theories of cause are similarly critically analysed. They include the possibility of an abnormal and inappropriate accumulation of growth factors in the sphincter or a diminution of the muscle relaxing chemical nitric oxide.
The evidence that there is a primary genetic contribution to the cause is also critically analysed.
Introduction
It all started with a Scotsman. Patrick Blair a surgeon apothecary from Perth, in 1717 reported the history and appearance at post mortem. “the child was 5 months old and was so emaciated that he appeared rather to have decreased, than to have increased, from the time of his birth; the whole body not weighing above 5 pounds----the pylorus and almost half the duodenum were cartilaginous and something akin to an ossification so that no nourishment could have passed into the intestines---“. The symptoms had been classical including that of a great appetite. No cause was proposed [1].
Surprisingly, it took some time before an abnormality in the stomach contents as the cause of PS was considered. Indeed, it was only in 1903 that Freund writing in a German journal found the hydrochloric acid content to be in excess of normal and proposed hyperacidity as the cause. Others, notable Berend in 1910, confirmed the findings and agreed. Moreover, in those early days, alkaline gastric infusions after stomach lavage were considered helpful in securing a cure. The associated reduced feeding was compensated by nutritious enemata. [2]
It is to Dr. John Thomson M.D. from Edinburgh in 1921 that we owe the understanding that that the tumour was indeed muscular hypertrophy; the condition sometimes self-cured; restricting feeds was important and, most importantly, minor forms were common and may self-cure from dietary restriction alone. [3] He further proposed that repeated sphincter contraction caused sphincter hypertrophy but did not specify what stimulated contraction. He based these opinions on a series of over 100 patients.
Primary Hyperacity Theory
For reasons obscured in the mists of time, the hyperacidity idea was not developed. An implied suspicion that the hyperacidity was due to retained acid behind a closed pylorus may have been the reason. The major interest understandably was the debate between the medical treatment involving atropine and immediate surgical cure.
This then was the position in 1951 when a meeting was convened at the Royal Society of Medicine, London. The subject was; Discussion on the treatment of Congenital Pyloric Stenosis.
All the big guns were there-, Denis Brown F.R.C.S., David Levi (by then a hero of 100 consecutive pyloromyotomies without mortality) and Dr. N.M. Jacoby, a specialist in both medical and surgical treatment. At that time Eumydrin-a variety of atropine absorbed directly from the buccal mucosa and suited for the vomiting baby was in fashion. Its effect in reducing acid secretion (and spasm) would be more secure. Here is a contribution from the floor of the meeting by Dr. Richard Bonham-Carter one year after graduating F.R.C.P.
“The pharmacology of Eumydrin is not completely understood. The
Pylorus is more responsive to changes of pH than to any other influence and I suggest that Eumydrin really acts indirectly by altering gastric secretion and hence the acidity of gastric juice. I wonder whether the line of treatment suggested by this idea had been explored”.
Another voice that of Dr. Harold Weller is also recorded.
“I have many times observed typical gastric peristalsis and projectile vomiting
In the first fortnight of life, and their disappearance under treatment with Eumydrin”. [4]
For this brief moment the mists had cleared and the sun was shining through. Sadly, no authoritative figure rallied to their support.
My personal involvement occurred simply by chance. Dr. Miller, an anaesthetist from Texas had documented in 1941 the phenomenon of a temporary wave of baby hyperacidity within a few hours of birth which lasted 1-2 days. [5] Before gastrin was discovered he thought that a chemical might pass from mother to baby and cause secretion of acid.
I read the paper and decided to measure gastrin in mothers and in babies.
We did not prove that gastrin transfer occurred- others have subsequently shown that it did in dogs [6] -but we did show that the day 4 gastrin was so much higher than the cord blood on Day 4 that it was higher than in fasting adults. [7] At the moment of birth, the infant stomach contains swallowed amniotic fluid and is neutral or alkaline. Since gastrin was rising at a time when we knew that acidity was also rising, it suggested that the negative feed-back between gastrin and antral acidity has not yet developed.
We followed this with a hypothesis about the cause of PS which relied on gastrin being secreted from a distended antrum due to milk feeds being unable to leave the stomach sufficiently quickly Gastrin stimulated acidity then continued the process of sphincter hypertrophy. We then compared fasting gastrin in PS babies and matched controls. There was no difference. [8]
What if the primary abnormality was instead an inherited hyperacidity? What if Dr. Freund, had been correct. It was already known that the greatest stimulus to sphincter contraction was acid entering the duodenum.
We measured the basal acid secretion.
The results were unequivocal. Acid secretion were increased in the PS group---volume; free acidity and total acidity. [9] These results have been confirmed by others. Hyperacidity has been found to persist even one week after a successful pyloromyotomy. [10]
Consequently, retained acid is not the reason.
When viewed from the perspective of inherited hyperacidity, astonishingly many of the clinical features were quickly explained.
Family history--- suggests a multi-factorial inheritance akin to acid inheritance.
Male predominance---premature male babies [11] and adult males secrete more acid than females and adults with duodenal ulcer share the same 5/1 male predominance as well as a predominance of blood Group O with PS babies. [12]
Pre-operative alkalosis -- When babies vomit and become alkalotic PS is invariably present13. Babies who vomit for other reasons are never alkalotic.
The acid model –for producing PS in puppy dogs involves artificially increasing acid secretion by pentagastrin injections. (Prof. Dodge’s model). [14]
Long term hyperacidity---studies reveal that in adulthood, PS babies suffer more frequently from hyperacidity problems. [15]
First -born phenomenon-- PS babies classically have a relish for feeds and it is easier for a novice mother to overfeed even when vomiting starts. Babies fed 3 hrly. Have a greater frequency of PS than babies fed 4 hrly. [16] The sphincter contracts most frequently and with greater amplitude in response to feeds [17]—and thus there is an explanation for the first-born phenomenon--and so it goes on!
There were two features which were yet unexplained. The presentation classically at 4 weeks (A) and the tendency to self-cure in the minor cases of PS. (B).
The insensitivity of the negative fed-back between gastrin and acidity has already been mentioned.
If the negative feed-back takes time to mature, a rise in gastrin and acidity would be expected from birth- and they do rise. [19]
Similarly, a temporary peak in acidity would be predicted-and Agunod’s curve shows that this also occurs.
Since gastrin secretion is unrestrained and maximally stimulated, no further gastrin increase would occur after feeds- and it does not. [20, 21, 22].
Similarly there is little difference between the maximally stimulated acid secretion and the fasting secretion in new born infants, testifying further to the lack of restraint by a falling gastrin. Acid is being stimulated already maximally within the potential of an immature gastric mucosa. [23]
The evidence supports the maturation of negative feed-back sometime after 24 days. [20, 21]A full account of the evidence in support of a temporary delay in establishing the negative feed-back has recently been published. [19]
This phenomenon explains the rise of gastrin and the rise of acid secretion soon after birth. It explains why the already hyperacid PS babies become critically hyperacid and develop hypertrophy. It explains why the combination of the early and later acidity protects the baby from enteric infections.
There is truly nothing new under the sun. In this particular topic, we have to look back to go forward.
Alternative Modern Theories.
Nitric Oxide (NO) deficiency.
In 1998 Prof. Furchgote was one of three researchers awarded the Nobel Prize for the discovery of NO, another new non-adrenergic non cholinergic (NANC) neurotransmitter. As with many great discoveries chance played a major part.
Transverse or spiral strips of rabbit aortic muscle were exposed to chemicals to see if contraction occurred.
Normally acetyl choline produced contraction. Occasionally, and most frequently with the transverse strips, a sudden relaxation was observed. The transverse strips histologically were found to contain to contain more preserved endothelium than the spiral strips. The spiral strip assistant had been rubbing the endothelium off in his rougher preparation!
Further studies discovered that the preserved endothelial factor, the Endothelial Derived Relaxing Factor (EDRF) was the cause of the muscle relaxation. EDRF was subsequently found to be the dissolved gas nitric oxide (NO) and its sub-endothelial location was important in controlling blood pressure through yet another negative feed-back mechanism [24]. NO caused an immediate but short-term relaxation of NANC innervated gut muscle [25]. But was a deficiency of NO implicated in the pathogenesis of PS?
Deficiencies of a NO intermediary, nitric oxide synthetase (NOS) have been reported in the sphincter muscle of some PS babies [26, 27]. Others have documented gastric stasis and sometimes pyloric hypertrophy in mice in which the NOS gene has been knocked out [27, 29]. In 1996 Chung and others, while conceding that the inheritance is multifactorial, produced evidence to support nucleotide variability for the promoter region of the neuronal NOS gene in the families of PS babies [30]. In 2009 Lagerstedt and others from the Karolinska Institute in a bigger analysis of 82 babies found no such association [31].
A specific genetic contribution has never been established. Recent genetic analyses have simply confirmed that heterogeneous multifactorial genetic inheritance is the norm [32]. The concordance rate in monozygotic twins while greater than that in dizygotes, is still only between 0.25 and 0.44 [33].
Others have reported abnormalities within the pyloric stenosis sphincter muscle of various growth factors. The truth is that for both sphincter growth factors and for local concentrations of NOS, there has never been, or ever could be, satisfactory ethical controls. The controls for the growth factor studies have exclusively come from post-mortem controls-infants up to 20 hours after death [34]. NOS studies have also used controls consisting of post-mortem controls or babies requiring pyloroplasty up to 13 years of age [26]. What would happen to the other NANC innervated muscles in the gut if these reported abnormalities were actually true?
These observations clearly do not stand up to critical analysis.
In any event when a muscle repeatedly contracts, as demonstrated famously by the great 18th. Century anatomist John Hunter, it becomes hypertrophic. It does so by attracting growth factors [35]. There is no evidence at all to support inappropriate accumulation of growth factors as a primary process in causing sphincter hypertrophy.
Similarly, a sphincter becoming hypertrophic from overwork has no need of the relaxing properties of NO. Even if there were reduced quantities common sense suggests that this would be because the sphincter is repeatedly contracting and seldom relaxed.
Neither the NO theory nor an abnormal accumulation of growth factors fits well with the essential time-sensitive progression or improvement of the condition. Such phenomena require to be connected to a changing environmental state which, in the case of the primary hyperacidity theory, is a developmental temporary peak acidity.
Acid secretion like many other human functions has a multifactorial inheritance. The inheritance of PS is classically multifactorial [36]. A genetic study into the inheritance of the capacity to secrete acid-the parietal cell mass- would perhaps be more appropriate and throw more light on the genesis of this condition.
Conclusion
There are three contemporary theories about the cause of Pyloric Stenosis of Infancy.
Primary Hyperacidity, abnormal accumulation of sphincter factors which encourage growth and a genetic abnormality.
Only the Primary Hyperacidity theory engages with the need to explain all the curious clinical features of this condition.
It is an aphorism which is undoubtedly true-listen to the patient-he is telling you the diagnosis.
In the case of Pyloric Stenosis of Infancy in the absence of speech-observe the patient, he (less likely to be she) is directing you to the cause.
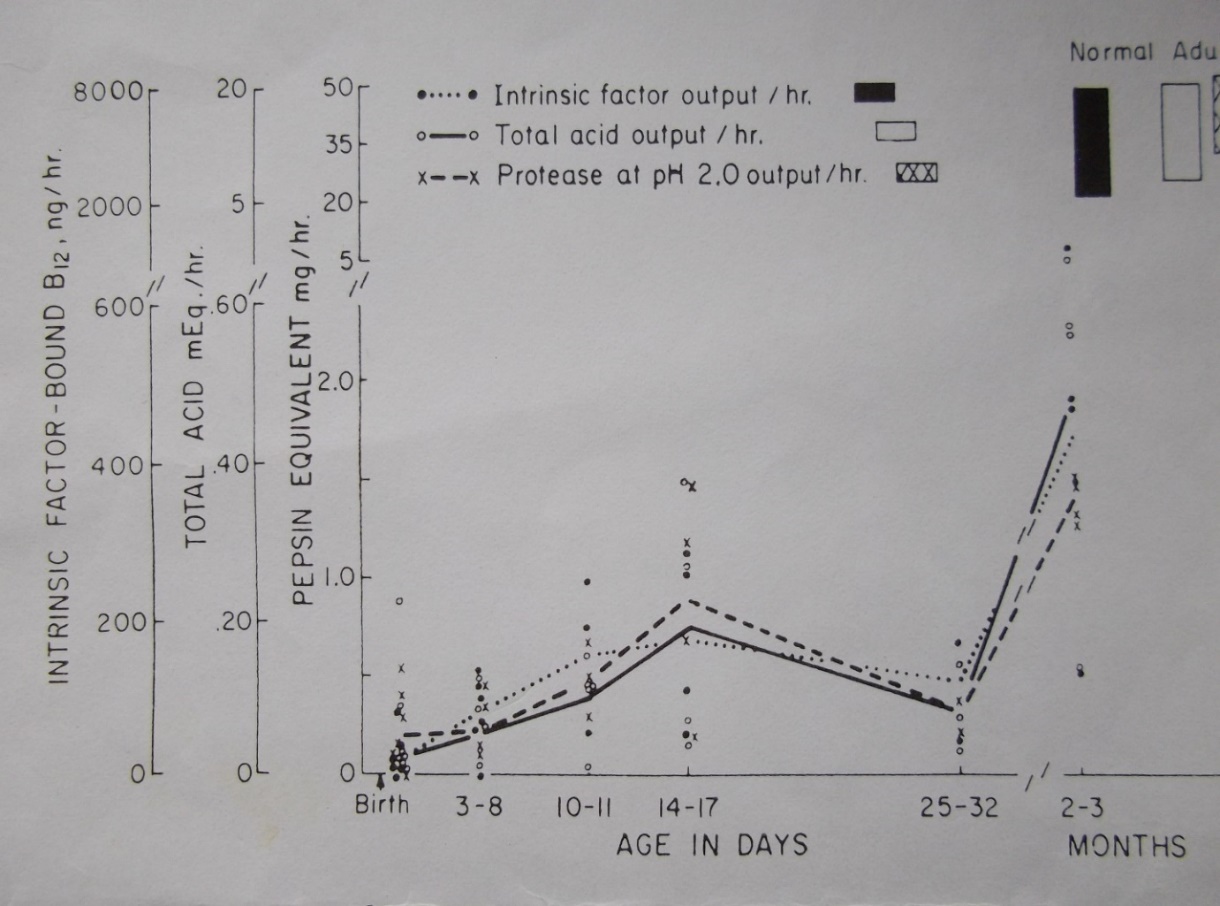
Figure 1. Total acid output in normal development peaks between 14-17 days and thereafter falls. Proteose (pepsin) and intrinsic factor follow the same pattern.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org