Pediatrics and Child Health Issues
OPEN ACCESS | Volume 6 - Issue 1 - 2026
ISSN No: 2836-2802 | Journal DOI: 10.61148/2836-2802/JPCHI

Malathi Sathiyasekaran 1*, R. Ganesh 2,
1Senior Consultant Pediatric Gastroenterologist, Rainbow Children’s Hospital, Chennai, TamilNadu, India
2Senior Consultant Pediatrician, Rainbow Children’s Hospital, 157, Anna Salai, Guindy, Chennai-600015,TamilNadu, India
*Corresponding author: Malathi Sathiyasekaran, Senior Consultant Pediatric Gastroenterologist, Rainbow Children’s Hospital, 157, Anna Salai, Guindy, Chennai-600015,TamilNadu, India
Received date: December 26, 2020
Accepted date: December 29, 2020
Published date: December 31, 2020
Citation: Sathiyasekaran M, R. Ganesh. “Approach to Metabolic Liver Disease in Children.’’. Pediatrics and Child Health Issues, 1(1); DOI: http;//doi.org/03.2020/1.1002.
Copyright: © 2020 Nigusu Derese. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly Cited.
Metabolic liver diseases (MLD) is a collective term for inherited single enzyme defects of inborn errors of metabolism (IEM ) which primarily or secondarily involve the liver. With recent advances in diagnosis and therapeutics, most inherited metabolic liver disorders are now being diagnosed and treated. This article outlines the important metabolic liver diseases in children and highlights the approach to evaluation and management.
Introduction:
Metabolic liver diseases (MLD) is a collective term for inherited single enzyme defects of inborn errors of metabolism (IEM ) which primarily or secondarily involve the liver. Since liver plays a pivotal role in several anabolic and catabolic biochemical reactions involving carbohydrates, protein, lipid and minerals, it is the target organ in several IEM. Hepatomegaly, hepatosplenomegaly and / or disturbed liver function or structure form an integral part of this disorder. There are more than 700 disparate IEMs that present with liver disease in infancy and childhood [1]. however the clinical manifestations are limited. MLD are generally considered a rare subset of liver diseases not amenable to treatment .However with advances in diagnosis inclusive of immuno-histochemistry staining in pathology, enzyme estimations, gas chromatography–mass spectrometry in blood/urine and genetic studies it is possible to make a definite diagnosis. A parallel advancement has also occurred in therapeutics in the form of special dietary formulae, enzyme replacement therapy, novel medications , hepatocyte and liver transplantation and gene therapy which has made a positive change in the outlook of MLD. Many inherited metabolic liver disorders are treatable and therefore performing appropriate investigations, making a definitive diagnosis and initiating prompt treatment are essential to ensure a good outcome. This article outlines the important metabolic liver diseases in children and discusses the approach to evaluation and management .
Pathophysiology:
The pathophysiology of MLD appears very complex and is diagramatically represented in Figure 1.
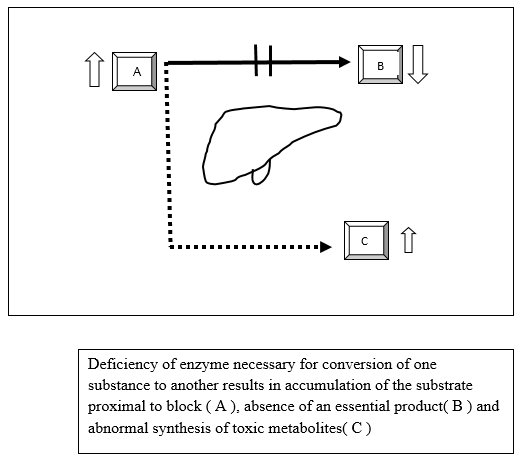
Figure 1 : Schematic representation of pathogenesis of Metabolic liver disease
The metabolic block could leads to organ dysfunction either due to a downstream deficiency of a metabolic product (B) such as glucose in GSD or an upstream excess of the metabolic substrate (A) such as glycogen or an alternative pathway (C) resulting in accumulation of toxic metabolites(tyrosinemia,galactosemia). The disorders can be classified pathophysiologically into three main groups [2]:
1.Disorders that involve the synthesis or catabolism of complex molecules eg: lysosomal and peroxisomal disorders. The partly degraded molecules accumulate in the affected organs, including the liver and disrupts the organ function.
2.Disorders, of energy metabolism caused by a deficiency in energy production and its consequences eg glycogen storage disorders [GSD], fatty acid oxidation and mitochondrial disorders.
3.Disorders caused by the accumulation of toxic compounds arising as a consequence of the enzyme defect, as occurs in galactosemia and tyrosinemia type I.
Incidence of MLD: In India 25000 newborns are born annually with MLD and account for 14 % of childhood liver disease. [3] The incidence of each MLD may vary geographically. Metabolic liver disease constitutes 13-43% of acute liver failure in young children and 5-20% in older children [4]. MLD presenting as chronic liver disease may vary from 10-40 % [5] and 4-12% of neonatal cholestasis syndrome [6].
Age of presentation: MLD can present at any age [ from prenatal, neonatal, infancy to adolescence and even adulthood coinciding with the peaks of catabolism. From the therapeutic point of view, the MLDs can be divided into four-time frames e.g., neonatal age, during infections, puberty and pregnancy. The MLDs differ in the age of presentation and the etiological conditions may vary from centre to centre.
Manifestations of MLD; The manifestation of MLD depends on the age of onset, substrate involved, duration of disease and degree of hepatocyte or other organ injury. The diagnosis is often delayed as the symptoms may be intermitten tand in the interval period, the patient may be free of clinical or biochemical abnormalities. The altered metabolic environment may affect a single organ or have systemic effects. The organs that have high metabolic rate and /or high energy requirement such as the brain, liver kidney heart and those with a high turnover of cell products such as the bone marrow or the growing skeleton are most susceptible.
Liver involvement usually arises as a primary effect of the IEM itself ;however it may vary from i) No involvement ( Gilbert's syndrome),ii) Normal liver with other organ involvement Crigler Najjar Syndrome (brain ),primary hyperoxaluria ( kidney ).iii) Mild liver involvement with more involvement of other organs,urea cycle defects and organic acidurias,iv) Slow liver damage GSD I and GSD III. v)Significant liver injury in infancy and children ( Galactosemia,Tyrosinemia, Wilson disease)
Inheritance :The majority of MLDs are inherited as autosomal recessive though autosomal dominant ( Acute Intermittent Porphyria ), X linked [Ornithine transcarbamylase (OTC) deficiency]or from maternal mitochondrial DNA ( Mitochondrial hepatopathies) can also occur.
Approach to MLD:
I.History: A detailed history as shown in (Table I ) may point towards a diagnosis of MLD. Since MLD is a genetic disorder, the history should extend to include gestation and also extended family members. Children born to mothers with Acute Fatty Liver Pregnancy or HELLP (Hemolysis Elevated Liver Enzymes Low Platelet) should be followed up for Fatty Acid Oxidation defect. Similarly, if there is a history of Intra hepatic Cholestasis of Pregnancy, there is a possibility of PFIC in the child. Galactosemia should be suspected in a neonate with recurrent vomiting. Poor scholastic performance in a child may be a clue for Wilson Disease. Dietary history includes symptoms which occur after ingestion of fructose rich foods such as honey, fruit juices, syrups as in fructose intolerance. Aversion to protein rich foods points towards urea cycle disorders. Craving for protein rich foods and aversion to carbohydrates towards citrin deficiency.
|
Clues in history |
Clues in History |
|
Positive family history of liver disease |
Recurrent episodes of similar illness at times of catabolic stress(fever ,exercise ,prolonged fasting ) |
|
Unexplained sibling deaths |
Episodes of unexplained metabolic derangement or hypoglycemia |
|
Recurrent fetal loss,abortions, AFLP and HELLP during pregnancy |
H/o change or avoidance of specific diet or amount of intake before illness |
|
Very few males in the family could be due to a X-linked disorder |
Sudden Infant Death Syndrome and psychiatric illnesses |
|
H/o parental consanguinity |
H/o abnormal growth or development |
|
Unusual body odours |
Microcephaly, macrocephaly, coarse features, mental retardation |
|
Acute neonatal illness |
FTT, rickets, dysmorphic features |
|
Presence of unexplained Coma |
Cataracts, Impaired vision |
|
Developmental delay, Unexplained convulsions, hypotonia, hypertonia |
Unexplained hepatomegaly, hepatosplenomegaly, elevated serum transaminases, recurrent jaundice |
Table I: Clues in history and examination to suspect MLD
II. Physical examination : Assessment of growth and development is essential in all MLDs. A thorough examination is an important step in evaluation before doing sophisticated tests[ Table II].
|
Physical Features |
Disorder |
|
Facial dysmorphism |
Mucopolysaccharidoses and peroxisomal disorders |
|
Doll like facies, Chubby cheeks[Figure 6) |
Glycogen storage disorder I and citrin deficiency |
|
Inverted nipples ,Abnormal fat pads |
Congenital disorder of glycosylation Ia,b |
|
Extensive mongolian spots |
GM1 gangliosidoses |
|
Hypopigmented, thin, brittle, kinky hair |
Menkys kinky hair disease |
|
Cataract |
Galactosemia, Wilson disease |
|
Cherry red spots |
Niemann pick disease, Tay Sach disease |
|
Supranuclear vertical gaze palsy |
Gaucher disease type 3, Niemann Pick C Disease |
|
Scratch marks |
Chronic cholestatic disorders,PFIC ,BASD |
|
Fractures |
Chronic cholestasis ,Gaucher disease |
|
Rickets |
WD, HFI, Fanconi Bickel syndrome ,Tyrosinemia |
|
Skeletal dysplasia (dysostosis multiplex), large skull,spinal deformities, and short, thick tubular bones. |
Mucolipidosis ,Mucopolysaccharidosis |
|
Reye like illness |
LCHAD,FAO |
|
Neurodevelopmental regression |
Mucolipid disorder |
|
Multisystem disease |
Mitochondrial hepatopathy |
Table II : Clues in general physical examination to suspect MLD
The phrase " the eyes sees what the mind knows " is very true in MLD. Apart from the abdominal examination a meticulous neurological, cardiac, respiratory assessment is essential. Ophthalmological examination should be included in the protocol and performed by an experienced opthalmologist for cataract, KF ring [Figure 2], cherry red spot and retinal pimentary changes.
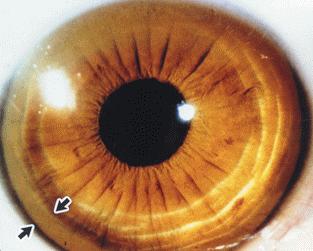
Figure 2.Kayser Fleicher Ring
Clinical Presentation and Common causes of MLDs : Many MLD present in the neonatal period and early infancy but some may manifest later in childhood .
|
Presentation |
Disorder |
|
i.Hydrops fetalis: Neonatal ascites and non immune hydrops fetalis |
Gaucher disease, Niemann Pick disease, Barth syndrome, GM 1 gangliosidoses, congenital disorders of glycosylation (CDG) and glycogen storage disease type IV |
|
ii.Neonatal Cholestasis Syndrome [NCS] |
Tyrosinemia, galactosemia, bile salt transport disorders (PFIC1-6), Bile acid synthesis defects, Niemann–Pick disease type C,peroxisomal disorders,, citrin deficiency, and Carbohydrate Disorders of Glycosylation |
|
iii.Acute Liver Failure |
Tyrosinemia type I, galactosemia, mitochondrial hepatopathy ,respiratory chain disorders and congenital disorders of glycosylation |
|
iv.Hyperbilirubinemia |
Criggler Najjar [CN] I and II |
|
v.Organomegaly |
Isolated hepatomegaly is seen in Wolman Disease , Glycogen Storage Disorder, Fatty Acid Oxidation defect and Citrin deficiency. Significant Splenomegaly is seen in Niemann Pick Disorder |
|
vi.Others /normal liver |
Organic acidemia and urea cycle disorder |
Table III. MLD in neonatal period and infancy
II.MLD in older children : There are 6 common presentations in older children(Table IV) .Three common MLD seen in older children are :
|
Presentation |
Disorder |
|
1.Isolated hepatomegaly |
GSD I ,Hereditary fructose intolerance and Fatty Acid Oxidation Test |
|
II.Hepatosplenomegaly/ splenohepatomegaly |
Glycogen storage disease III,IV, Wilson disease, Gaucher’s disease, Niemann Pick disease & Mucopolysaccharidosis |
|
III.Chronic Liver Disease |
Wilson disease, Gaucher disease, GSD IV, Hereditary Fructose Intolerance, Cystic fibrosis and Tyrosinemia |
|
IV.Acute Liver Failure [ALF]and Acute on Chronic Liver Failure [ACLF] |
Wilson disease ,Bile salt transport defects, PFIC 3 |
|
V. Recurrent Jaundice |
Crigler Najjar syndrome II, Gilbert’s syndrome, Dubin Johnson syndrome and Rotor Syndrome |
|
VI.Chronic Cholestasis |
Progressive Familial Intrahepatic Cholestasis and Inborn Error of Bile Acid Synthesis |
Table IV.MLD in older children
A)Glycogen Storage Disorder: These are a group of AR inherited disorders due to enzyme defect in metabolism of glycogen resulting in increased accumulation of normal or abnormal glycogen in various tissues. Liver is most severely affected, in addition skeletal muscle, heart, kidney, bones and brain may be involved.There are more 13 types of which type I (Glucose-6-phosphatase deficiency) and III (Amylo-1,6 – glucosidase deficiency) are more common. In India GSD account for 8-24 % of MLD3.In GSD I, the features are massive hepatomegaly, hypoglycemia, growth retardation, early morning seizures, renomegaly and mild elevation of transaminases. In GSD III, some distinct features are hepatosplenomegaly, elevated transaminases, muscle involvement in the form of cardiomyopathy or myopathy. Liver biopsy shows swollen hepatocytes with PAS positive diastase sensitive glycogen .Fibrosis may be seen in GSD III. Genetic studies help to confirm the diagnosis. Uncooked corn starch is very effective .Liver transplantation may the definitive treatment for GSD III.
B)Wilson Disease: This is an AR inherited treatable disorder of copper metabolism due to mutations in ATP7B gene located on chromosome13 and is a common MLD worldwide. In India, it accounts for 13 % of pediatric liver disease and 33-53 % of MLD3. The defective pType ATPase interferes with transport of copper from the liver, resulting in abnormal accumulation of copper in various tissues, liver kidney, brain, RBC. WD should be suspected in any child more than 3 years with liver disease. The classic triad of low ceruloplasmin, KF ring( Figure 2) and elevated 24 hr urine copper is diagnostic but may not be present .Liver biopsy may show steatosis ,glycogen nuclear vacuolation and increased copper content in dry weight. Genetic studies are confirmatory .D Pencillamine and Trientene should be given life long .Liver transplantation is indicated in the presence of acute liver failure or end stage liver disease.
C) Progressive Familial Intrahepatic Cholestasis (PFIC): PFIC is a heterogenous group of autosomal recessive inherited disorders of bile salt transport and excretion resulting in hepatocellular cholestasis presenting usually in infancy and early childhood or rarely later in life and progresses to end stage liver disease and death. PFIC constitutes 10 to 15 % of cholestasis in children7 and 10-15% of liver transplants8. PFIC 1 ,2 and 3 are the most common among the six types of PFIC. which occur due to specific mutations resulting in functional deficiencies of canalicular proteins responsible for transport of bile salts. Pruritus is a predominat symptom(Figure 3) and progression to cirrhosis is universal. Low gamma glutamyl transpeptidase is an important surrogate marker seen in all PFICs except PFIC 3. Liver histopathology with immuno-histochemistry may support however the confirmatory diagnosis is by doing genetic analysis.The response to choleretics is variable .Partial internal or external biliary drainage relieves pruritus in PFIC 1 and PFIC2.Liver transplantation is the definitive therapy for PFIC 2 PFIC3 and PFIC4. [9]
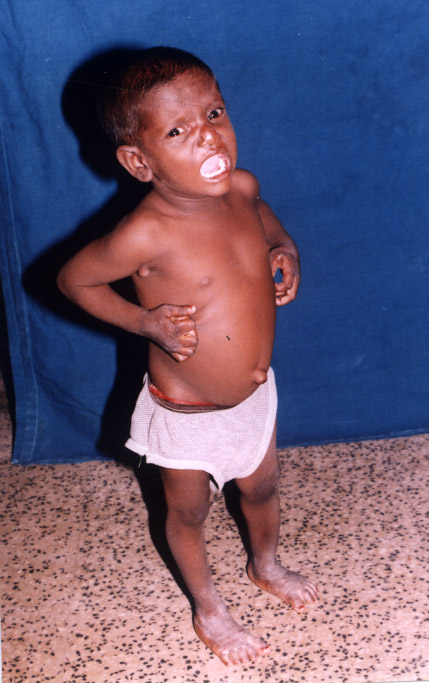
Figure 3: Child with intractable pruritus PFIC /BASD
III. Investigations: Since it is difficult to distinguish clearly between MLD from non IEM liver disorders, it is advisable to include tests for the common and treatable metabolic conditions ( eg Wilson disease) when assessing liver dysfunction due to any cause.
The investigations for MLD comprise of A. Baseline investigations, B. Basic metabolic investigations and C. Advanced metabolic investigations including molecular genetic testing.
A.Basic investigations:
1.Complete blood count and peripheral smear : Anemia may be a feature of Wolman and Gaucher disease. Acanthocytosis on smear is suggestive of Abetalipoproteinemia. vacuolated lymphocytes in Wolman disease or Lysosomal disorders.
2.Liver function tests: Though these tests may be abnormal in several non MLDs it may help in diagnosis of some MLD. Normal or low GGT levels are seen in progressive familial intrahepatic cholestasis 1 to 6 (except PFIC 3 ) and in bile acid synthesis defects ( BASD ) and thus low GGT is an excellent marker to diagnose a subset of metabolic cholestatic disorders.
3.Serum Bile acids(BA): Bile acids are disproportionately increased in inherited Bile salt transport defects PFIC 1 to 6 but decreased in BASD. This helps us to differentiate PFIC and BASD since GTP is low or normal in both these conditions.
4.Serum Uric acid: High levels of serum uric acid points towards Lesch Nyhan syndrome and low levels point towards xanthine/hypoxanthine disorders and Molybdenum cofactor deficiency.
5.Serum Ceruloplasmin( Cp ): It is useful as a screening test for Wilson Disease. The normal value is 20-40 mg /dl . A value < 10 mg /dl is diagnostic of WD.
6.Serum iron and serum Ferritin: Levels are increased in hemochromatosis.
7.Lipid profile: Abnormal lipid profile points towards glycogen storage disorders, lipid storage disorders Wolman disease and Nieman Pick disease.
8.Creatinine Phosphokinase : Levels are increased in GSD III with muscle involvement and mitochondrial disorders.
10.Serum Alphafetoprotein : A high AFP in the absence of hepatic malignancy is a sensitive marker for tyrosinemia.
2 Urine : Reducing substance for glucose may indicate diabetes but Non Glucose Reducing Substance is seen in Galactosemia ,fructosemia.
3.Imaging
i)Xray :Adrenal calcification is seen in Wolman disease[Figure 4], epiphyseal stippling is seen in Zellweger syndrome.
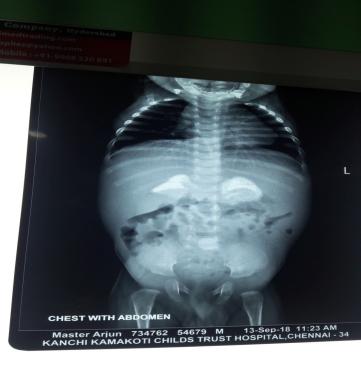
Figure 4: Xray abdomen adrenal calcification in Wolman disease
ii) USG abdomen may show the changes seen in chronic liver disease such as coarse echotexture of liver, ascites and dilated portal vein or collaterals.
4.Bone marrow examination: Presence of large macrophages with crinkled paper cytoplasm is seen in Gaucher disease and foamy cells in Niemann Pick disease[ Figure 5].
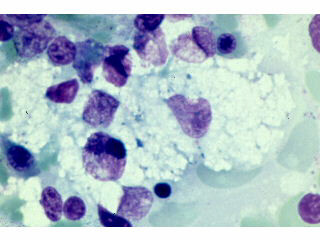
Figure 5 : Bone marrow showing large foamy cell in NP disease
5.Liver biopsy: Though an invasive test, liver biopsy may reveal some characteristic changes as seen in GSD I and III [Figure 6],
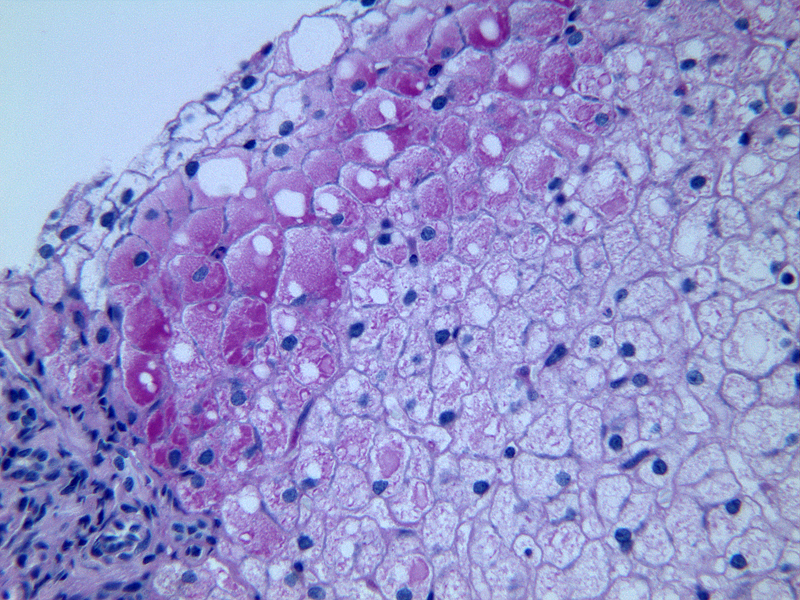
Figure 6: Liver biopsy showing swollen hepatocytes with glyco gen PAS +ve
the characteristic "fructose holes” in hereditary fructose intolerance, nuclear vacuolation and steatosis with positivity for copper in Wilson disease, bland cholestasis and Byler's bile on Electron microscopy in PFIC I and steatosis in cystic fibrosis ,fatty acid oxidation defect and citrin deficiency.
B. Basic Metabolic investigations--.Initial investigations should include tests for the more common metabolic conditions according to the presenting features. GALAK is a good mnemonic to remember the tests which help in diagnosis of MLD associated with significant biochemical derangements.
i)Glucose: Hypoglycemia - defined as blood glucose levels less than 40 mg/dL (plasma glucose levels < 45mg/dL) is seen in glycogen storage disorders, gluconeogenesis defects, galactosemia, fatty acid oxidation disorders and congenital disorders of glycosylation
ii)Ammonia: Ammonia estimation should be done from a free flowing blood sample obtained from either arterial or venous puncture and the normal values are <100 μmol/L (neonates), <80 μmol/L (infants) and <50 μmol/L (older children). Hyperammonemia is seen in urea cycle disorders, organic acidemias, liver failure
iii) Lactate: Lactate estimation should also be done from a free flowing blood sample .The normal values are 0.2-2.5 mmol/L. Increased lactate levels are seen in poor perfusion states, organic acidemias, fatty acid oxidation disorders, defects in pyruvate metabolism and electron chain defects.
iv)Acid base status: Acidosis is defined as pH less than 7.3 .Metabolic acidosis is seen in organic acidemias, defects in pyruvate metabolism, mitochondrial disorders. Alkalosis is seen in urea cycle disorders.
v)Ketones: Ketonuria, an increase in the urinary excretion of ketones points to organic acidemia. Ketonuria in fed and fasting states can occur in ketone body handling disorders such as succinyl-CoA transferase (SCOT) deficiency or beta-ketothiolase (BKT) deficiency.
C.Advanced investigations: The combination of history,clinical examination ,base line and basic metabolic investigations helps to narrow down the differential diagnosis of MLD to a great extent. The advanced metabolic investigations can now be tailored and selected to confirm diagnosis according to the provisional diagnosis(TableV).
|
Probable MLD diagnosis |
Confirmatory tests for diagnosis |
Treatment |
|
Hereditary Fructose intolerance |
Mutation analysis |
Elimination of fructose, sucrose and sorbitol from the die |
|
Gaucher disease |
Glucocerebrosidase assay (leukocytes or cultured fibroblasts). Mutation analysis |
Enzyme replacement therapy[Imiglucerase] Substrate reduction therapy with miglustat, eliglustat |
|
Fatty Acid Oxidation |
Plasma acyl carnitine assay |
Avoiding fasting, Carnitine |
|
Wolman disease |
Acid lipase assay (leukocytes or cultured fibroblasts) and/ or DNA sequencing |
Enzyme replacement therapy[sebelipase alfa] |
|
Mitochondrial disorders |
Mutation analysis for known mitochondrial and nuclear DNA defects |
Supportive therapy |
|
Organic Acidemia |
Urine organic acids. Plasma Acylcarnitine.Gene testing |
Special formulas |
|
Urea Cycle disorders |
Plasma amino acids assay Urine orotic acid Gene testing |
Special formulas |
|
Zelleweger |
Mutational analysis |
Supportive treatment |
|
Bile Acid Synthetic Defect |
Mutational analysis |
Supportive therapy,choloretics, Vitamin supplements, antipruritic agents, Biliary diversion procedures, Liver transplant |
|
Crigler Najjar |
Mutational analysis |
Phenobarbitone, phototherapy, liver/hepatocyte transplant |
|
Cystic Fibrosis |
Mutational analysis |
Supportive treatment |
Table V : Advanced Metabolic tests for diagnosis and specific treatment in MLD
Special methods of specimen collection, storage, and transport may be necessary, and it is best to plan such investigations in consultation with a specialist metabolic laboratory before collection.
i)Molecular gene testing: Genetic testing by next generation sequencing in metabolic liver diseases is important for accuracy of diagnosis to provide information on prognosis, for cascade carrier testing and for prenatal or pre-transplantation diagnosis. Whole exon sequencing (WES) that covers exons and intronic regions of the gene is widely available . WES helps us to arrive at the diagnosis. WES that covers the enitre coding and non-coding areas of the gene is very expensive. Clinical exome serquencing is useful when the WES is not feasible. Both biochemical tests and NGS are complimentary to each other.
Treatment : The treatment of MLD range from modification of diet in certain conditions like galactosemia, hereditary fructose intolerance and glycogen storage disorders, special enteral formulas in aminoacidopathies,organic acidemia and urea cycle disorders. Specific medications in Crigler Najjar syndrome type II, Tyrosinemia and Wilson disease are recommended. Enzyme replacement and substrate reduction therapies in lysosomal storage disorders .( Table V)
Hepatocyte Transplantation : Hepatocyte transplantation has been used in treating Crigler-Najjar syndrome type I, GSD type 1a and UCD.
Liver transplant ; LT is indicated in MLD where it will reverse or arrest the disease progression.The common MLDs where LT has changed the natural history of the disease are Tyrosinemia ,wilson disease,GSD IV. LT is contraindicated in disease with severe multisystemic involvement e.g., mitochondrial defects.
Dual Liver and Kidney transplant : In Primary Oxaluria liver and kidney transplant may be warranted.
Gene therapy.Liver-targeted gene therapy with rAAV8 vectors has efficaciously corrected the glycogen storage of GSD Ia and Pompe disease in preclinical studies and may be the answer in future. [10]
Bone Marrow Transplant :In Wolman disease BMT has been attempted with good results.
The treatment also includes supportive therapy with choleretics in cholestatic disorders, Vitamin supplementation, approriate management of hepatic encephalopathy, blood products and vitamin K in coagulopathies and appropriate recognition and treatment of complications.
Prevention: The preventive components in MLDs include genetic counseling, sibling screening and antenatal diagnosis .Immunisation with Hepatitis B and A vaccines is essential to prevent worsening of underlying disease.
Key Messages.
Funding source: None
Financial disclosure: None
Conflict of Interest: None
Contributors:
RG reviewed literature and drafted manuscript. MS drafted manuscript and reviewed manuscript for intellectual content. MS will act as the guarantor.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org