Gupta Ashutosh1*, Aneja Anjila2, Bahl Neena3, Arora Rupam4, Nadir Lovelenna5 and Saini Pankaj6
*1Head- Foetal Medicine & Medical Genetics, Artemis Hospitals, Sector 51, Gurgaon, India.
2Director & Head - Department of Minimal Access Surgery (Gynecology), Fortis Memorial Research Institute, Gurgaon, India.
3Director - Department of Minimal Access Surgery (Gynecology), Fortis Memorial Research Institute, Gurgaon, India.
4Senior Consultant Obstetrics & Gynecology, Max Balaji Hospital, Cloudnine Hospital, Patparganj,New Delhi, India.
5Senior Consultant - Obstetrics & Gynecology, Artemis Health Institute, Fortis La Femme S 549 Greater Kailash New Delhi, India.
6Senior Consultant - Department of Radiology, Manipal Hospitals, Dwarka, Sector 6, New Delhi, India.
*Corresponding Author: Gupta Ashutosh, Head- Foetal Medicine & Medical Genetics, Artemis Hospitals, Sector 51, Gurgaon, India.
Received date: December 01, 2023
Accepted date: December 29, 2023
Published date: January 05, 2024
Citation: Gupta Ashutosh, Aneja Anjila, Bahl Neena, Arora Rupam, Nadir Lovelenna and Saini Pankaj. (2024). “Prenatal ADPKD – Does Mutation analysis helps?”. International J of Clinical Gynaecology and Obstetrics, 4(1); DOI: 10.61148/2836-0737/IJCGO/028.
Copyright: © 2024 Gupta Ashutosh. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disease; a late-onset multiorgan disorder affecting kidneys, liver and blood vessels of brain. Renal insufficiency manifests at around 4-5th decade of life and around half of them have end-stage kidney disease (ESKD) by 6th decade of life.
Presumptive prenatal diagnosis of bilateral ADPKD was made at 14 weeks of gestation; confirmation of the diagnosis was made by follow up 18 weeks ultrasound and by genetic molecular mutation analysis by gene sequencing.
As both of the parents showed no ultrasound findings of ADPKD, prenatal ADPKD was presumed to be de-novo.
Genetic analysis revealed missense (non-truncating) mutation; even though it was helpful in the diagnosis but didn’t shed any light or give headway regarding the future course of pregnancy. The pertinent questions remain unanswered like; future course, most likely prognosis, timeline for end stage renal disease, renal dialysis or need for or time of renal transplant.
Prediction regarding the future course is similar to the non-invasive ultrasound diagnosis.
Literature suggests that ADPKD detected antenatally and genetic truncating mutations might be associated with fulminant or severe course of the disease, but is still ambiguous regarding the decision making for the continuation of pregnancy.
ADPKD; prenatal; confirmation; genetic; sequencing; missense; prognosis; fulminant
Introduction:
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disease; a late-onset multiorgan disorder affecting kidneys, liver and blood vessels of brain. It is characterized by multiple cysts in both kidneys, cysts in liver, intracranial aneurysms, cysts in pancreatic, seminal vesicles and arachnoid membrane.
Renal disorder may include early-onset hypertension, kidney pain, and renal insufficiency manifesting in 4th decade of life, with majority to have end-stage kidney disease (ESKD) by 6th decade. Liver cysts mat be more common in females, ADPKD carriers have 5 folds increased risk of intracranial aneurysm than the general population.
ADPKD demonstrates substantial clinical variability both renal and extra-renal manifestations. ADPKD is diagnosed by age-specific renal imaging criteria; family history of either an affected first-degree relative or a heterozygous pathogenic variant in PKD1, PKD2 or or digenic ADPKD mutations which is found in a minority of families. Most affected individuals have an affected parent, however around 10-20% of the affected individuals have a de novo mutation.
Initial nephron and collecting duct formation is normal but there is later cystic dilatation of these structures leading to secondary loss of normal anatomy. [1, 2] Age-specific ultrasound criteria are established for the diagnosis of ADPKD, with majority of the affected individuals would have at least one renal cyst by the age of 30 years. [3, 4] Sonographic characteristics are well established; large hyperechogenic kidneys, loss of cortico-medullary differentiation (CMD), multiple cysts of variable size, associated hepatic, pancreatic or ovarian cysts. [4] Presence of three or more (unilateral or bilateral) kidney cysts in between 15-39 years of age, two or more cysts in the age group of 40-59 years; large echogenic kidneys without distinct macroscopic cysts in an infant or child who is at 50% risk for ADPKD. [5]
Fully penetrant (non-hypomorphic) biallelic pathogenic variants in either PKD1 or PKD2 gene is lethal and incompatible with life. [6,7] Authors have reported families with individuals homozygous for PKD1 pathogenic variants. [8] A hypomorphic allele in trans with a non-hypomorphic (fully penetrant) pathogenic variant has been identified in infants with prenatal-onset ADPKD. [9]
Biallelic hypomorphic variants may result in early-onset disease with an apparently negative family history and thus mistaken for autosomal recessive polycystic kidney disease (ARPKD). [10] Homozygosity of a hypomorphic PKD2 variant arising due to by uniparental disomy can also present as neonatal-onset ADPKD. [11] Double heterozygotes for a pathogenic variant in both PKD1 and PKD2 may present as more severe kidney disease than reported in heterozygotes. [12]
Majority of the affected individuals have a positive family history of ADPKD; however, de novo pathogenic variants may be found in around 10%-20% of cases.[13] Parental germline mosaicism may be responsible for 0.25% of families with ADPKD and have been reported. [14]
With the advanced ultrasound machines and technique, prenatal ADPKD are now more frequently being diagnosed, however prenatal in-utero cases have been reported to have a poor prognosis. [15]
Sonographers attempting to make prenatal diagnosis of ADPKD, must be aware of the subtle sonographic characteristics of ADPKD, as in-utero expression may be limited to kidneys being enlarged, hyperechogenic and but rarely grossly cystic [16] which is required for precise characterization of corticomedullary differentiation. However, authors have reported ultrasound to have good sensitivity for the diagnosis of ADPKD. [17, 18] Even-though, nomograms are available for the fetal kidney size; hyperechogenicity and corticomedullary differentiation (CMD) of the fetal kidneys is usually assessed only subjectively. [19, 20]
Echogenicity of the fetal kidney is assessed by comparing it with that of the adjacent liver and spleen. However, ultrasonic characteristics of the maternal abdominal wall, amniotic fluid volume, gain setting of the machine and frequency of the probes used influence hugely this most subjective part of the diagnosis. [19, 21]
Abnormal cilia structure, function or both may lead to abnormalities in cell proliferation and tubular differentiation leading to cyst formation. [22] Polycystin-1, Polycystin-2, fibrocystin and BBS8 are found in the primary cilia of renal epithelium, forming calcium-permeable ion channel complex, regulating the cell cycle and the function of the renal primary cilium, thus establishing that ciliary dysfunction leads to cyst development.
Molecular characterization helps in better understanding of the disease; but at least half of the families affected don’t have any informative genetic information and the affected or carrier family members are ascertained through the prenatal evaluation of the fetus.
Prenatal diagnosis of the pre-symptomatic condition has its own pitfalls and ethical issues to deal with. Firstly, ADPKD is an adult-onset disease, manifesting in fourth decade of life with highly variable clinical symptomatology. Presymptomatic diagnosis of the adult-onset disease with uncertain course and prognosis makes prenatal diagnosis and counselling very difficult for both the clinician and the patient.
Ultrasound and gene sequencing techniques has helped in better understanding of the disease and complex molecular characterization of the ADPKD, allowing prenatal diagnosis of ADPKD.
Case Report:
Primigravida in routine ultrasound (USG) examination at 14 weeks of gestation was detected to have bilateral enlarged and hyperechogenic kidneys (Fig1), amniotic fluid was within normal range with mild subcutaneous edema over the head and chest (Fig 2). There was no history of polycystic kidney disease in family, bilateral kidneys of both the parents were assessed but were radiologically and no evidence of ADPKD was found.
As the presumptive diagnosis of bilateral ADPKD was made at 14 weeks of gestation, the author knew its fallacy. So, the parents were counselled regarding the possibility of the fetus being affected with bilateral ADPKD; which warranted a follow up USG for corroboration of the ultrasound findings and confirmation of the diagnosis. Couple was very apprehensive regarding the diagnosis, possible ways for confirmation of the diagnosis, possible course, long-term prognosis and outcome for the fetus and any need for renal dialysis or renal transplantation in postnatal life.
Follow up USG at 18 weeks, showed almost similar renal findings, enlarged hyperechogenic bilateral kidneys with accentuated corticomedullary differentiation and confirming the diagnosis of ADPKD (Fig 3) with mild to moderate subcutaneous edema over the head and thoracic cavity which still persisted (Fig 4).
As both the parental kidneys showed no ultrasound signs of ADPKD it was presumed to be de-novo. Parents decided for invasive testing (amniocentesis) and genetic analysis for the molecular confirmation of ADPKD.
After pre-test counselling parents were informed that fetal exome study might not answer all the questions being raised; like the effect of some (non-truncating) mutations in any one of the gene PKD1 or PKD2, degree of fetal renal affection and probable age of renal dysfunction, might not reflect on the necessity or time line for renal dialysis or transplant in postnatal life.
After informed proper consent, amniocentesis was done and fetal DNA obtained was processed for exome testing.
Exome study revealed the fetus to be compound heterozygous for the missense mutation (non-truncating) in PKD1 gene; c.11453G>T in Exon 41; likely pathogenic in nature and the second missense mutation c.7928G>T in Exon 21 of the PKD1 gene which was variant of uncertain significance (Fig 5).
Non truncating (missense) mutations are different from truncating ones. Even though genetic testing helped the molecular confirmation of the diagnosis but they didn’t answer the basic question whether to continue or take an irreversible decision with the pregnancy. Prediction regarding the future course is similar to the non-invasive ultrasound diagnosis. Truncating mutations might be associated with fulminant or severe course of the disease and might be helpful for decision making regarding the pregnancy.
Fetal gestational age was 22 weeks by the time molecular diagnosis was done, however the ultrasound findings were almost the same but were more explicit (Fig 6) and the fetus still showed submandibular edema with subcutaneous edema over the head (Fig 7).
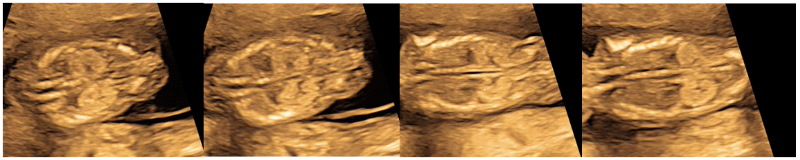 Figure 1: Ultrasound at 14 weeks of fetal gestation showing enlarged, hyperechogenic bilateral kidneys with normal amniotic fluid suspecting ADPKD.
Figure 1: Ultrasound at 14 weeks of fetal gestation showing enlarged, hyperechogenic bilateral kidneys with normal amniotic fluid suspecting ADPKD.
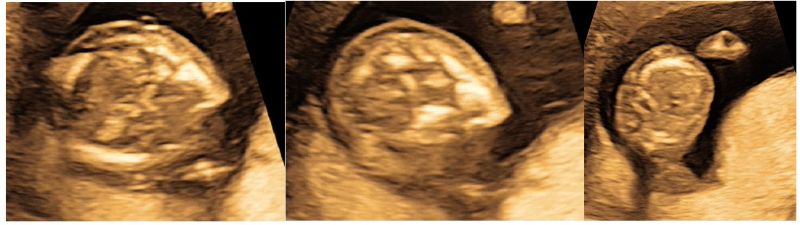 Figure 2: Ultrasound at 14 weeks of fetal gestation showing mildly increased subcutaneous edema over the head and upto thoracic cavity.
Figure 2: Ultrasound at 14 weeks of fetal gestation showing mildly increased subcutaneous edema over the head and upto thoracic cavity.
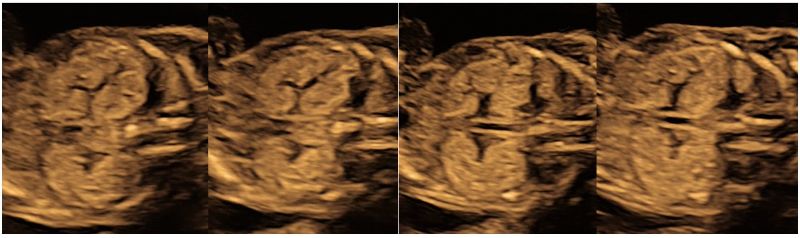 Figure 3: Ultrasound at 18 weeks shows similar renal findings of both enlarged, hyperechogenic kidneys with increased corticomedullary differentiation with normal amniotic fluid confirming the diagnosis of ADPKD.
Figure 3: Ultrasound at 18 weeks shows similar renal findings of both enlarged, hyperechogenic kidneys with increased corticomedullary differentiation with normal amniotic fluid confirming the diagnosis of ADPKD.
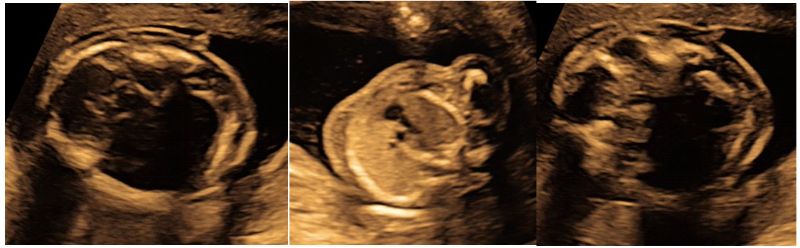 Figure 4: Ultrasound at 18 weeks of fetal gestation showing persistence of mildly increased subcutaneous edema over the head and upto thoracic cavity.
Figure 4: Ultrasound at 18 weeks of fetal gestation showing persistence of mildly increased subcutaneous edema over the head and upto thoracic cavity.
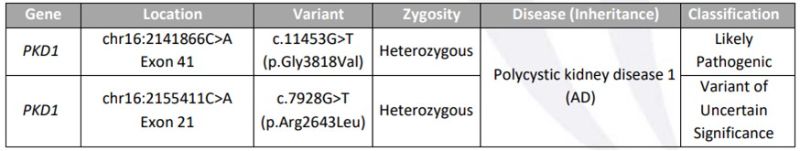 Figure 5: Showing the molecular characterization of the fetal exome study; Compound heterozygous with one likely pathogenic mutation, both of them however missense mutations (non-truncating mutation)
Figure 5: Showing the molecular characterization of the fetal exome study; Compound heterozygous with one likely pathogenic mutation, both of them however missense mutations (non-truncating mutation)
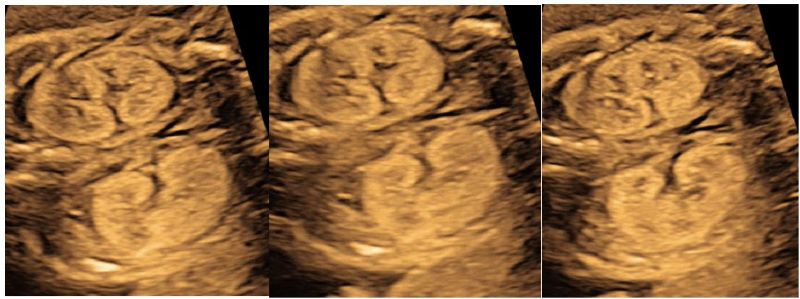 Figure 6: Follow up fetal ultrasound at 22 weeks of fetal gestation showing similar renal findings of both enlarged, hyperechogenic kidneys with explicit corticomedullary differentiation concording with the diagnosis of ADPKD.
Figure 6: Follow up fetal ultrasound at 22 weeks of fetal gestation showing similar renal findings of both enlarged, hyperechogenic kidneys with explicit corticomedullary differentiation concording with the diagnosis of ADPKD.
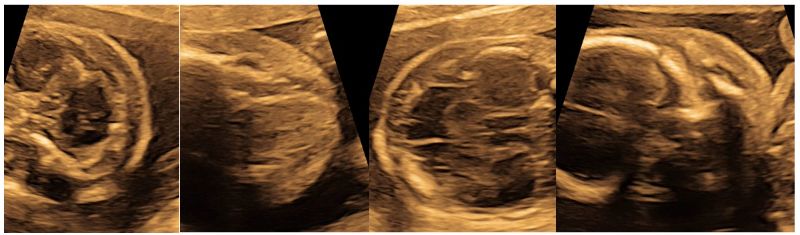 Figure 7: Ultrasound at 22 weeks of fetal gestation showing persistent mildly increased subcutaneous edema over the head and some edema in submandibular cavity which is a new entity.
Figure 7: Ultrasound at 22 weeks of fetal gestation showing persistent mildly increased subcutaneous edema over the head and some edema in submandibular cavity which is a new entity.
Discussion:
Isolated large and hyperechogenic fetal kidneys is a finding which might be difficult to comprehend by the sonographers. First, one cannot be certain of the diagnosis and second of the underlying pathology and how to explain the findings and possible consequences to the parents-to-be. Genetics is a complex and multifaceted specialty and more so when it comes to prenatal diagnosis.
Pre-conceptional genetic counselling is very complex which is in fact more the rule than the exception. Counselling can be straightforward if there is an index case (proband) in the family; difficult if there is none. In almost half of the cases of large hyperechogenic kidneys; this fetal feature ‘introduces’ this concept to the family leading to diagnosis of ADPKD in one of the asymptomatic parents, thus parents being diagnosed through the fetus.
Major impetus during the detailed fetal examination is on to state whether both the kidneys are present, collecting system is dilated and quantification of the amount of amniotic fluid. The ultrasonic texture and CMD of the kidneys are often neglected thus missing the probable diagnosis of ADPKD.
ADPKD is a common hereditary kidney disease; with incidence reported to be 1/1000. Clinical onset of the disease typically occurs in the fourth decade, however early onset during prenatal period or childhood has been reported. [23, 24] Prenatal cases of ADPKD have been reported with large hyperechogenic kidneys as the most common finding. [15, 23, 25] Prenatal diagnosis is now-a-days being performed by DNA analysis on chorionic villi at 11–13 weeks. [15, 26, 27] (Table No 1)
Hyperechogenic kidneys are now being increasingly diagnosed during the prenatal period. This might be reflective of kidney diseases with very different aetiologies, prognoses and outcomes, including obstructive dysplasia, bilateral multicystic dysplasia, genetically inherited renal disease ARPKD, ADPKD, genetic syndromes, Perlman syndrome, Beckwith–Wiedemann syndrome, Bardet–Biedl syndrome, Meckel syndrome, nephroblastomatosis, renal vein thrombosis, toxic injury, infections especially cytomegalovirus [29] ischemia, aneuploidy, and in some cases normal variants [19]
|
Cases |
Increased echogenicity |
Renal enlargement |
Renal cysts |
Family history |
Reference |
|
3 |
+ |
+ |
- |
- |
[15] |
|
5 |
+ |
+ |
- |
+ |
[23] |
|
3 |
+ |
+ |
- |
+ |
[28] |
|
Our Case |
+ |
+ |
- |
- |
|
Table 1: Characterization of Prenatal ADPKD
Conclusion:
It is really intriguing and interesting to know how the family’s reaction and response to the prenatal diagnosis of ADPKD is immensely affected by their previous knowledge of the disease (renal failure) and various forms of therapy (dialysis or transplantation) available. Final decision of the consultand (parents) also depends hugely on the individual perception of the severity of the disease. [30]
Isolated enlarged hyperechogenic with or without cystic changes in the fetal kidneys poses a significant diagnostic dilemma when discovered as an incidental finding during routine fetal ultrasound examination. The duality between the probability of the prenatal diagnosis and the late and uncertain prognosis of fetal ADPKD often makes one uncomfortable in presenting the diagnosis to the parents. There may be hugely diverse aetiologies with equally heterogenous and variable prognosis for the affected fetus.
Literature is now suggesting of strong evidence to support primary ciliopathy serving as a common pathway for progressive cystogenesis in the various forms of PKD. Molecular characterization of the disease allows for better understanding; identification of the defective gene in PKD family allows for precise genetic diagnosis in the fetus, especially when there is no index case in the family.
However, the diagnosis of prenatal ADPKD is ultrasound dependent, genetic molecular testing might help in confirming the diagnosis, but the basic question remains unanswered.
Mutations could be non-truncating (missense) or truncating; phenotypic effect of both of them shall be different. Even though they help in the diagnosis but they don’t give headway whether to continue or take an irreversible decision. Future course, likely prognosis, timeline for end stage renal disease, renal dialysis or need for or time of renal transplant remains unanswered. Prediction regarding the future course is similar to the non-invasive ultrasound diagnosis. Authors have reported that the genetic etiology can be elucidated in about 50% of the enlarged hyperechogenic kidneys, when it is excluded short term renal outcome is normal but the long-term outcome is uncertain. [31] Literature suggests that ADPKD detected antenatally and genetic truncating mutations might be associated with fulminant or severe course of the disease, but is still ambiguous regarding the decision making for the continuation of pregnancy.

Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org