
N.M. Zaikova, E.D. Gordeychuk, S.L. Morozov, S.E. Ryabova, V.V. Dlin*, A.A. Martynov
Veltischev Research and Clinical Institute for Pediatrics and Рediatric surgery of the Pirogov Russian National Research Medical University, Moscow, Russia.
*Corresponding authors: Vladimir Dlin, Veltischev Research and Clinical Institute for Pediatrics and Рediatric surgery of the Pirogov Russian National Research Medical University, Moscow, Russia.
Received Date: June 27, 2023
Accepted Date: July 14, 2023
Published Date: July 20, 2023
Citation: N.M. Zaikova, E.D. Gordeychuk, S.L. Morozov, S.E. Ryabova, V.V. Dlin, A.A. Martynov (2023). “Сlinical polymorphism of Barakat syndrome”. Clinical Research and Clinical Case Reports, 4(1); DOI: http;//doi.org/07.2023/1.1066.
Copyright: © 2023 Vladimir Dlin. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Barakat Syndrome (MIM#146255) is a rare autosomal dominant disease caused by GATA3 gene mutation and manifested by hypoparathyroidism (H), sensorineural deafness (D), and renal disease (R). HDR syndrome characterized by high clinical variability and prognosis. The exact prevalence of this disease is unknown, 180 cases are reported in the literature. Below are 2 clinical cases. De novo heterozygous mutation in the gene GATA3 was detected in both patients. Our observations demonstrate an antenatal presentation of GATA3-related cystic kidney diseases, variability of clinical phenotypes and different kidney prognosis of patients with Barakat Syndrome. The syndrome should be suspected in cases of early high-grade deafness and kidney disease presentation for the purpose of early diagnosis and appropriate therapy including the prevention of CKD progression.
Introduction
The Barakat syndrome ([OMIM] (146255) was first described by Barakat A.Y. et al. in 1977 in two brothers and a sister presenting with hypoparathyroidism, sensorineural deafness, and renal abnormalities [1]. Barakat syndrome, also known as HDR syndrome (hypoparathyroidism, sensorineural deafness, and various renal tissue abnormalities), is an autosomal dominant genetic disorder caused by haploinsufficiency of the GATA-binding protein 3 (GATA3) gene [2, 3]. The GATA3 gene is expressed in developing parathyroid glands, inner ear, kidneys, thymus, and central nervous system [3]. GATA3 plays a key role in T-cell development; in fact, GATA1, GATA2, and GATA3 are essential factors for haematopoiesis. GATA3, a trans-acting transcription factor specific to T-cells, is a protein belonging to the GATA family of transcription factors encoded by the GATA3 gene located on chromosome 10 in humans. The protein's polypeptide chain has a length of 443 amino acids, and its molecular weight is 47,916 (Figure 1) [4].
Fig. 1. Three-dimensional structure of the human GATE 3-Zf1 gene based on the mouse GATE 1 Nf1 model (Catherine W. Gaynor, 2009) [4]
Genetic variations that can cause HDR syndrome include missense or nonsense pathogenic variants, small insertions or deletions, and large deletions that lead to structural variations in the GATA3 gene [5]. Currently, there is no precise data on the prevalence of the disorder in the population because identifiable GATA3 variants are not present in all patients with clinical features similar to Barakat syndrome [6].
The Aim: To identify the variability of clinical manifestations in Barakat syndrome based on different pathogenic mutations
Materials and methods
An evaluation of the patients' medical history, physical examinations, laboratory tests, and molecular genetic research results was conducted. Protein-coding gene sequencing in humans using paired-end sequencing was performed with targeted enrichment of genomic DNA. The sequencing data were analyzed using an automated algorithm, which involved assessing sequencing quality parameters (FASTQC module), adapter and low-quality sequence removal (SEQPURGE module), alignment of reads to the hg19 human genome version (BWA MEM module), removal of optical and PCR duplicates (SAMBLASTER module), local alignment optimization (ABRA2 module), variant detection and quality-based filtration (FREEBAYES package), and variant annotation against clinical information databases (ENSEMBL-VEP module). The algorithm was tested on exome data with available "gold standard" genome decoding (Genome in a Bottle data). The algorithm demonstrated a sensitivity of 98.6% and an average specificity of 99.1%. Informed consent from the legal guardians of the children was obtained for their examination and description. Validation of the GATA3 gene was performed on both patients and their parents by Sanger sequencing (trio), confirming the mutation in this gene in both probands and absence of the mutation in the parents. The absence of this mutation in other family members suggests that it is a de novo mutation.
Clinical Case 1: A male child born in 2008 has been under the care of an otolaryngologist-audiologist since birth due to severe sensorineural hearing loss (4th degree) and cochlear implantation. The child has also been seen by an ophthalmologist for hypermetropia and astigmatism. There is no family history of intellectual disability, hypocalcaemia, renal insufficiency, or deafness. The child has been monitored by a paediatrician for bilateral kidney dysplasia since birth, along with persistent hypocalcaemia from 3 months of age. The child is from a non-consanguineous marriage and is the product of the mother's first pregnancy, which resulted in premature delivery at 35-36 weeks of gestation. At birth, the child had below-average height (46 cm) and weight (2400 g). Prenatal ultrasound examination at 16 weeks of gestation suggested the possibility of agenesis of one kidney. The child's physical development has been delayed, and there has been noted speech development delay.
The child was first examined at the RCI for P and PS nephrology department at the age of 13 due to elevated blood creatinine levels detected during a routine visit to the paediatrician. According to the clinical examination, the child has a very high physical development (height >97‰, body weight 90-97‰). Mild hypermetropia, hypermetropic astigmatism, equinus-varus foot positioning, flexion knee contractures, extension hip contractures, flexion finger contractures, and secondary gait abnormalities are noted. Laboratory investigations revealed hypocalcaemia (total calcium - 2.07 mmol/L, ionized calcium - 0.96 mmol/L), hyperphosphatemia (2.45 mmol/L), magnesium levels at the lower limit of normal, and low parathyroid hormone (PTH) levels (7.1 pg/mL). Kidney function is impaired, with a blood creatinine level of 93 mmol/L, blood urea level of 6.6 mmol/L, estimated glomerular filtration rate (eGFR) of 73.3 mL/min/1.73m², corresponding to stage 2 chronic kidney disease (CKD), and compensated acid-base status. The urinary syndrome is characterized by low molecular weight proteinuria (5-fold increase in beta-2-microglobulin), with no evidence of hypo/hypercalciuria or microalbuminuria. Audiogram findings indicate severe bilateral sensorineural hearing loss (grade 4) (Figure 2a). Renal ultrasound reveals differentiated renal parenchyma, with the right side showing poor differentiation and the left side showing inadequate clarity, thickened parenchyma, and increased echogenicity (slightly higher than liver echogenicity). Doppler ultrasound demonstrates diffuse depletion of blood flow in the renal cortex, and both kidneys show a few cysts with maximum dimensions of 0.4 x 0.3 cm on the right side and 0.3 x 0.3 cm on the left side. The kidney volume is reduced bilaterally, measuring less than 25‰ (Figure 2b).
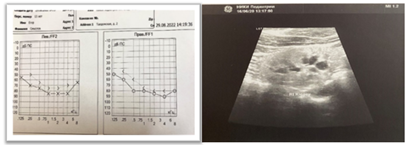
a) b)
Fig.2. a) Audiogram of patient E. 13 years old, b) Ultrasound picture of the right kidney of patient E, 13 years old
According to 24-hour blood pressure monitoring, there are no signs of arterial hypertension or hypotension. Densitometry results show a moderate increase in overall bone mineral density.
The child underwent molecular genetic testing, specifically whole-exome sequencing, which revealed a previously described pathogenic variant (rs1832792205) in a heterozygous state in exon 4 (out of 6) of the GATA3 gene. This variant leads to an amino acid substitution of arginine to glutamine at position 276 (p.Arg276Gln), classified as a missense mutation. Functional analysis demonstrated that the presence of this variant results in protein dysfunction. This mutation was confirmed by Sanger sequencing in the proband and was not detected in the parents (Table 1).
|
Gene |
Associated disease (OMIM) |
DNA alteration (Protein alteration) |
Zygosity (Type of inheritance) |
Frequency |
Classification of pathogenicity |
|
GATA3 |
Barakat syndrome (hypoparathyroidism, sensorineural deafness, and renal dysplasia) (146255; AD) |
chr10:g.8106004G>A ENST00000379328.3: c.827G>A ENSP00000368632.3: p.Arg276Gln |
Heterozygote |
0 |
Pathogenic |
Table 1. Results of molecular genetic examination of patient E, 13 years old
Based on the medical history and molecular genetic testing, the diagnosis of Barakat syndrome has been confirmed in the boy.
In order to provide renal protection, the boy has been prescribed enalapril at a dose of 0.2 mg/kg, and dynamic monitoring will be conducted.
Clinical Case 2: A 7-year-old boy (born in 2015) is under the care of a nephrologist due to congenital kidney abnormalities (bilateral kidney dysplasia, bilateral grade 3 vesicoureteral reflux corrected by endoscopy), and rapid progression CKD to stage 4 (hyperazotemia since 1 month of age, bilateral sensorineural hearing loss grade 2). The child is the product of the mother's first pregnancy, born at 40 weeks of gestation. The pregnancy was unfavorable, with persistent toxemia throughout. At birth, the child weighed 4150 g, measured 55 cm in length, and had Apgar scores of 7-8. The family history is significant, with the father having renal hypoplasia and kidney cysts. There is no consanguinity in the marriage. The child's early physical and neuro-psychological development was age-appropriate, and vaccinations were administered according to an individual schedule. The child has experienced frequent respiratory tract infections, recurrent urinary tract infections, and secondary obstructive recurrent pyelonephritis. According to the clinical examination, the child's physical development is above average and harmonious (height 75-90‰, body weight 90-97‰). The child is also being monitored by an otolaryngologist-audiologist for bilateral sensorineural hearing loss grade 2 (Figure 3a).
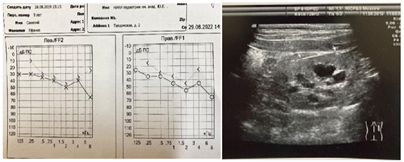
a) b)
Fig.3. a) Audiogram of patient C, 7 years old, b) Ultrasound picture of kidneys in patient C, 7 years old
Laboratory investigations revealed no hypocalcemia or hyperphosphatemia (total calcium - 2.3 mmol/L, ionized calcium - 1.17 mmol/L, phosphorus - 1.56 mmol/L). Hyperparathyroidism was noted, indicated by elevated parathyroid hormone (PTH) levels (122.4 pg/mL). The level of β-2 microglobulin (β-2 MG) was increased threefold, and there was moderate microalbuminuria (150 μg/mg creatinine). Impaired renal nitrogen excretion function was observed (blood urea nitrogen - 17.1 mmol/L, creatinine - 209 μmol/L), with an estimated glomerular filtration rate (eGFR) of 22.5 mL/min/1.73m2. The acid-base status showed subcompensated metabolic acidosis (blood pH 7.36, base excess (b) - 4.9 mmol/L, standard bicarbonate (HCO3 std) 21 mmol/L, partial pressure of carbon dioxide (pCO2) 34 mmHg).
According to ultrasound examination (Ultrasonography), the kidneys showed poor differentiation from surrounding tissues, with right kidney mobility of 5.3% and left kidney mobility of 3.7% (normal range up to 1.8%). Kidney rotation in orthostasis was observed, and the contour of both kidneys was uneven, with reduced size (right: 6x4.2x4.2 cm, volume 36.9 cm3; left: 5.7x4.5x4.5 cm, volume 48.3 cm3). The kidney volume was 10‰, and the parenchyma was undifferentiated with increased echogenicity (higher than liver echogenicity) and thinning at the poles of both kidneys. High-frequency linear ultrasound imaging revealed punctate hyperechoic inclusions in the renal pyramids. The renal blood flow was diffusely depleted, and the central echo complex was deformed. Bilateral caliectasis and megaureters were observed. There was hypoplasia with cysts in both kidneys. The child had undergone endoscopic correction of bilateral vesicoureteral reflux (Figure 3b).
Ambulatory blood pressure monitoring (ABPM) revealed stable systolic-diastolic arterial hypertension during night time.
Molecular genetic testing was performed on the child, and the results identified a previously undescribed, likely pathogenic variant in a heterozygous state in exon 4 (out of 5) of the GATA3 gene, leading to disruption of the canonical splicing site. This mutation was confirmed by Sanger sequencing in the proband and was not detected in the parents (Table 2).
|
Gene |
Associated disease (OMIM) |
DNA alteration (Protein alteration) |
Zygosity (Type of inheritance) |
Frequency |
Classification of pathogenicity |
|
GATA3 |
Hypoparathyroidism in combination with sensorineural deafness, and renal dysplasia (146255) |
10:g.8106102G>A ENST00000379328.3: c.924+1G>A |
Heterozygote (Dominant) |
0 |
Probably pathogenic variant |
Table 2. Results of molecular genetic examination of patient S, 7 years old
Considering the medical history, presence of hypoplasia of both kidneys, bilateral vesicoureteral reflux, and impaired renal nitrogen excretion function (stage 3b chronic kidney disease), along with the findings from molecular genetic testing, the diagnosis of Barakat syndrome has been established. The secondary hyperparathyroidism is attributed to the development of renal insufficiency following the bilateral endoscopic correction of vesicoureteral reflux, leading to secondary contracted kidneys.
Currently, due to stage 4 chronic kidney disease, the child is receiving metabolic and symptomatic therapy, nephroprotection, and undergoing vaccination as per the kidney transplant preparation program.
Discussion.
The exact prevalence of Barakat syndrome is unknown, as the HDR syndrome is highly heterogeneous [7-10]. To date, approximately 180 patients have been reported from the United States, Japan, India, China, Europe, and the Middle East. The prevalence of the disease is similar among different ethnic groups, genders, and age groups. The number of individuals with HDR syndrome is expected to increase as clinical awareness of this condition grows [9].
The typical clinical triad of Barakat syndrome includes primary hypoparathyroidism, sensorineural deafness, and renal anomalies. It is the haploinsufficiency of developmental genes that leads to a wide range of penetrance and, consequently, the clinical manifestations of the syndrome [11]. The triad of hypoparathyroidism, sensorineural deafness, and renal insufficiency is usually observed in 62.3% of patients, while 28.6% of patients present with hypoparathyroidism and deafness only, and approximately 2.6% of patients have deafness and kidney disease without hypoparathyroidism [12,13]. A wide spectrum of phenotypic variability has been described in other studies published in the scientific literature (Figure 4) [7, 10, 11, 13-15].
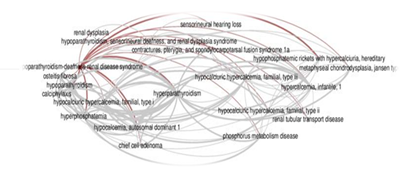
Fig. 4. Graphical network of 20 major diseases associated with HDR syndrome (Nesbit MA, 2004) [16].
In our study, both boys were found to have sensorineural hearing loss and renal dysplasia with cysts, and only in the first case was hypoparathyroidism present. It was in this patient that a pathogenic missense mutation was detected in a heterozygous state in exon 4 (out of 6) of the GATA3 gene, resulting in the substitution of arginine with glutamine at position 276 (p.Arg276Gln), which causes the complete triad of Barakat syndrome. In the second patient, a previously undescribed potentially pathogenic variant was identified by molecular genetic analysis in a heterozygous state in exon 4 (out of 5) of the GATA3 gene, leading to disruption of the canonical splice site. This variant manifested as an incomplete Barakat syndrome (renal dysplasia with cysts and sensorineural hearing loss of grade 2) without hypoparathyroidism. However, renal hypoplasia and cysts were detected in his father. It is possible that the renal malformation in the father is not associated with the GATA3 gene mutation, but the presence of mosaic somatic GATA3 mutation in the kidneys of the father cannot be ruled out. Thus, we have demonstrated the variability of clinical manifestations of Barakat syndrome depending on the gene mutation.
The most consistent feature of Barakat syndrome is bilateral sensorineural hearing loss, which typically presents in childhood [14, 17]. Patients develop moderate to severe sensorineural hearing impairment at early stages, predominantly bilateral, symmetric, and worsening at the higher frequencies [14]. It is known that high-frequency sensorineural hearing impairments in individuals with Barakat syndrome progressively worsen with age [12, 18]. Both boys in our case had hearing impairment since birth. However, despite sensorineural hearing loss being reported in the literature, the exact onset time is not well established, as it is a slowly progressive condition, and most patients do not seek early medical attention. During clinical examination, if a patient has profound or obvious hearing loss or there is a family history of deafness, it may provide a clue to the early diagnosis of HDR syndrome. If mild or moderate hearing loss is not detected during routine clinical examination, and the patient is also unaware of its presence, the diagnosis of HDR syndrome becomes uncertain. This represents the "gray zone" of the disease [7, 10, 14].
Hypoparathyroidism in HDR syndrome can range from asymptomatic to presenting with myalgia, neuromuscular excitability disorders, non-febrile seizures, or severe tetany due to profound hypocalcemia [10, 19, 20]. Initially, most cases of HDR were primarily treated as primary hypoparathyroidism [9, 18]. In the first patient, hypoparathyroidism with marked hypocalcemia was detected without signs of tetany, while the second patient had hyperparathyroidism with a normal blood calcium level. Neither of our patients exhibited neurological symptoms with seizure syndrome.
Renal manifestations occur in 90% of cases of Barakat syndrome and represent the most heterogeneous clinical component. They can be functional or structural and include nephrotic syndrome, cystic kidney, renal dysplasia, hypoplasia or aplasia, pelvicalyceal deformity, vesicoureteral reflux, and nephrosclerosis, which were observed in our patients [18, 21]. There are also reports of proteinuria, hematuria, renal tubular acidosis, and nephrocalcinosis in patients with Barakat syndrome [8]. In our patients, the renal syndrome was characterized by low molecular weight proteinuria and microalbuminuria. The prognosis for patients with Barakat syndrome usually depends on the severity of renal insufficiency [17, 22]. The literature describes progressive progression of CKD in most patients, leading to the development of end-stage renal disease requiring renal replacement therapy. Patient #2 in our case exhibited signs of renal insufficiency at an early stage.
Therefore, only one of our described patients (case 1) presented with the complete triad of Barakat syndrome (hypoparathyroidism, sensorineural hearing loss, and cystic dysplasia of both kidneys). The absence of the mutation in the family members suggests a de novo mutation, although it is typically reported that Barakat syndrome cases are inherited dominantly [10, 23]. We compared our clinical cases with previously described cases of Barakat syndrome reported in the literature (Table 3) [10, 12, 17, 19, 20, 22].
|
Characteristic |
Clinical cases |
Akie Nakamura et al. (2011) [18] |
Nasrollah Maleki et al. (2013) [21] |
Liu C, et al. (2015) [12] |
Gül Yesiltepe Mutlu et al. (2015) [10] |
Xue-Ying Chu et al. (2017) [19] |
Tetsuji Okawa et al. (2015) [16] |
|||||
|
Case 1 |
Case 2 |
Patient 1 |
Patient 2 |
Patient 3 |
Patient |
Patient 5 |
Patient 6 |
Patient 7 |
Patient 8 |
Patient 9 |
Patient 10 |
|
|
Sex m/f |
m |
m |
f |
f |
f |
f |
f |
f |
f |
f |
f |
f |
|
Age of diagnosis |
13 years |
7 years |
1 month |
11 months |
2 months |
1 month |
13 years |
58 years |
19 years |
13 years |
14 years |
33 years |
|
The function of parathyroid gland at diagnosis time |
||||||||||||
|
Hypoparatheriosis +/- |
+ |
- |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
Clinical features |
low weight gain |
hyperazotemia from 1 month of life |
low weight gain |
convulsions |
pain in the lower extremities |
convulsions |
convulsions |
convulsions |
convulsions |
convulsions |
convulsions |
convulsions |
|
Р (1.29-2.26 mmol/L) |
2.45 |
1.56 |
unknown |
2.907 |
unknown |
2.71 |
2.196 |
2.32 |
1.71 |
3.068 |
3.7 |
1.51 |
|
Ca Ionized (1.13-1.32 mmol/L ) |
0.96 |
1.17 |
1.2 |
1.1 |
unknown |
1.5 |
1.4 |
1.325 |
1.86 |
1.675 |
1.62 |
1.3 |
|
PTH (16-62 pg/ml) |
7.1 |
122,4 |
7–10 |
5–9 |
20 |
9 |
13 |
5 |
9.96 |
20 |
22 |
7 |
|
Sensorineural hearing loss |
4 deg |
2 deg |
2 deg |
2 deg |
2 deg |
2 deg |
2 deg |
4 deg |
2 deg |
2 deg |
2 deg |
2 deg |
|
Ultrasound signs |
cystic kidney dysplasia |
cystic kidney dysplasia, VUR |
norm |
norm |
norm |
renal dysplasia |
norm |
renal dysplasia |
hypoplasia of both kidneys |
hypoplasia of the left kidney |
norm |
hypoplasia of the right kidney |
|
Genetic research |
chr10:g.8106004G>A ENST00000379328.3: c.827G>A ENSP00000368632.3: p.Arg276Gln |
10:g.8106102G>A ENST00000379328.3: c.924+1G>A |
Exon 6 c.1063delC p.L355X |
Exon 3 c.432insG p.K303X |
Exon 4 c.784A > G p.R262G |
Intron 5/exon 6 boundary c.1051-1G > T p.1351fsX18 |
Exon 5 c.942 T > A p.C318S |
Not identified |
Exon 2 c.529dupC p.Arg177profsX126 |
Exon 4 p.R276Q c.827G > A |
Exon 2 c.286delT p.W96GfsX99 |
Exon 4 p.R299Q |
Table 1. Comparative phenotypic characteristics of presented clinical cases of children with HDR syndrome and cases previously described in literature
Conclusion.
Thus, despite having the same diagnosis confirmed by molecular-genetic methods, the prognosis for the lives of both patients with Barakat syndrome varies. Considering the earlier and more significant decline in GFR in the second patient (reaching stage 4 of CKD at the age of 7), the need for dialysis and probable kidney transplantation will arise much earlier compared to the first patient, significantly compromising their quality of life. However, it is worth noting that despite the considerably higher GFR in the first child, their degree of neurosensory hearing loss is more severe than that of the second child, which also affects the patient's quality of life. These presented data confirm the phenotypic heterogeneity of Barakat syndrome, manifested by varying degrees of neurosensory hearing loss, hypoparathyroidism, different anomalies of the urinary system, and varying degrees of progression of kidney failure. The earlier decline in GFR and progression of CKD lead, respectively, to an early need for renal replacement therapy, which determines the prognosis for the lives of each patient with Barakat syndrome. The differences in the frequency of detection or variability of the main symptoms and the early development of kidney failure detected in HDR syndrome justify the early conduct of molecular-genetic research. The timely diagnosis of Barakat syndrome will enable early initiation of nephroprotective therapy and reduce the rate of progression of CKD.
Conflict of interest:
The authors of this article confirmed the lack of conflict of interest and financial support, which should be reported.

Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org