Clinical Research and Clinical Case Reports
OPEN ACCESS | Volume 7 - Issue 1 - 2026
ISSN No: 2836-2667 | Journal DOI: 10.61148/2836-2667/CRCCR


Gudisa Bereda
Department of Pharmacy, Negelle Health Science College, Guji, Ethiopia
*Corresponding authors: Gudisa Bereda, Department of Pharmacy, Negelle Health Science College, Guji, Ethiopia.
Received: June 07, 2022
Accepted: June 16, 2022
Published: June 22, 2022
Citation: Gudisa Bereda (2022). “Clinical Pharmacology of Acetaminophen”. Clinical Research and Clinical Case Reports, 3(2); DOI: http;//doi.org/05.2022/1.1055.
Copyright: © 2022 Gudisa Bereda. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Paracetamol is used worldwide for its analgesic and antipyretic actions. It has a spectrum of action identical to that of non-steroidal anti-inflammatory drugs and specifically to look like the cyclooxygenase type 2 selective inhibitors. Acetaminophen is a safe, effective, well-tolerated and cheap analgesic and anti-pyretic medications with moderately few adverse effects when used at the recommended therapeutic dosage. Acetaminophen it suppresses cyclooxygenase type 1 and 2 through metabolism by the peroxidase work of these isoenzymes. Paracetamol lowers mild to moderate fever and pain by affecting the chemical messengers in the brain that regulate body temperature. With chronic coincident usage of paracetamol and zidovudine, neutropenia frequently happens and is presumably owing to the decreased metabolism of zidovudine.
Introduction
Paracetamol is a ubiquitous analgesic that acts as a competitive inhibitor of COX enzymes. It is metabolised in the liver via several pathways, enclosing glucuronidation and sulfation, to accelerate its excretion from the body. At therapeutic doses, comparatively 10% of paracetamol is metabolised by CYP450 enzymes to NAPQI, a greatly reactive, hepatotoxic compound. NAPQI is highly conjugated with glutathione to a non‐toxic metabolite for excretion. Although, with toxic doses of paracetamol, glucuronidation and sulfation pathways are become saturated, and glutathione deposits can become decreased. This sequence in the concentration of NAPQI causes hepatocellular detriment and potentially leading to acute liver failure [1-4]. Paracetamol is used worldwide for its analgesic and antipyretic actions. It has a spectrum of action identical to that of NSAIDs and especially to look like the COX-2 selective inhibitors. Paracetamol is on average a weaker analgesic than NSAIDs or COX-2 selective inhibitors, but it is frequently preferred because of its improvement gastric tolerance [5, 6]. Acetaminophen, a non-salicylate identical to ASA in analgesic potency, has substantiated efficacy and visible safety at all steps of pregnancy in criteria to therapeutic doses. Its settled safety profile for usage has been settled in a current survey of thousands of pregnant women, without increasing risks of congenital abnormalities or different adverse pregnancy consequences [7-10]. Paracetamol is an analgesic used to ameliorate pain and great fever. It is also used for treating headaches, muscle/joint pains, backaches, toothaches, and colds. One of the most effective remedies for decreasing body temperature, it commences functioning in an hour [11, 12].
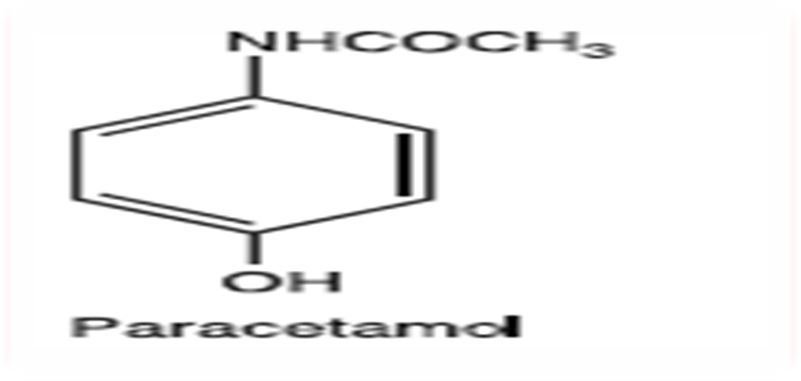
Figure 1: chemical structure of paracetamol
Paracetamol has chiefly anti-pyretic (decreasing the levels of prostaglandins in the hypothalamus) and analgesic properties; it does not interrupt with COX 2 and does not affect the different constituents of inflammation (swelling and redness). As paracetamol has no action on COX 1 at a therapeutic dose it has few side effects. The maximum recommended daily therapeutic dose of paracetamol for adults is 4g (8 x500mg tablets) [13-15]. Paracetamol is a safe, effective, well-tolerated and cheap analgesic and anti-pyretic medication with moderately few adverse effects when used at the recommended therapeutic dosage [16].
Mechanism of action: It acts by preventing prostaglandin generation by its action on COX-3 enzymes, (an optional splice product of cox-1 enzyme)/ despite the similarities to NSAIDs, the mode of action of paracetamol has been not completely clarified, but it is now generally accepted that it inhibits COX-1 and COX-2 through metabolism by the peroxidase function of these isoenzymes. Is a sequence in the inhibition of phenoxyl radical conformation from a critical tyrosine residue, indispensable for the activity of COX-1 and COX-2 and PG production. Paracetamol reveals selectivity for suppression of the generation of PGs and related factors, when less level of arachidonic acid and peroxides are applicable. Reversely, the drug shows less activity at substantial levels of arachidonic acid and peroxides. The sequence is that paracetamol does not prevent the severe inflammation of rheumatoid arthritis and acute gout, but it prevents the least inflammation sequencing from e.g. the extraction of teeth. Unlike both non-selective NSAIDs and selective COX-2 inhibitors, paracetamol inhibits different peroxidase enzymes involving myeloperoxidase. Suppression of myeloperoxidase includes the paracetamol oxidation and the concurrent decreased formation of halogenating oxidants (e.g. hypochlorous acid, hypobromous acid) that perhaps associated with multiple inflammatory pathologies involving atherosclerosis and rheumatic infirmities. Therefore, according to this mechanism, the development [17, 18]

Figure 2: mechanism of action of acetaminophen
Indications: Paracetamol lesser mild to moderate fever and pain by affecting the chemical messengers in the brain that control body temperature. It’s also combined with different pain-relief and anti-sickness mediations. Furthermore, its ingredient is section of a wide range of cold and flu remedies. It is broadly used for: Reducing fever; alleviating and relieving headaches; decrease pain caused by menstrual cramps; toothaches; backaches; decreasing pain caused by arthritis (specifically, osteoarthritis) in joints in the hands, knees, hips, colds etc [19-21].
Drug interactions: The speed of absorption of paracetamol perhaps increased by metoclopramide or domperidone and absorption decreased by cholestyramine [22]. The anticoagulant consequence of warfarin and different coumarins perhaps accelerated by extended daily use of paracetamol with increased risk of bleeding. Intermittently doses have no important consequence [23]. Paracetamol is extendedly metabolized in the liver and can therefore interact with medicinal products with the identical metabolic pathway or initiate/suppress the identical metabolic pathway [24, 25]. Chronic use of alcohol or medicinal products which initiate liver enzymes like rifampicin, barbiturates, certain anti-epileptic medications (e.g. carbamazepine, phenytoin, phenobarbital, and pirimidone) and St. John’s Wort can increase the hepatotoxicity of paracetamol as a sequence of an increased and hasty formation of toxic metabolites. Cautiousness is therefore compulsory with coincident usage of enzyme-inducing medications [26, 27]. Probenecide prevents the binding of paracetamol to glucuronic acid reducing paracetamol clearance by a factor of about 2. If probenecide is taken coincidentally the paracetamol dose should be decreased [28]. Paracetamol can increase the plasma accumulation of chloramphenicol [29]. With chronic coincident use of paracetamol and zidovudine, neutropenia frequently happens and is likely owing to the decreased metabolism of zidovudine [30]. Salicylamide perhaps extend the elimination half-life of paracetamol [31].
Dosage and Administration: Adults (involving the elderly) and children aged 12 years and over: Oral administration solely 500 mg paracetamol/65 mg caffeine to 1000 mg paracetamol/130 mg caffeine (1 or 2 tablets) every 4 to 6 hours as necessitated [32]. Maximum daily dose: 4000 mg/520 mg (paracetamol/caffeine), and do not exceed the stated dose. The lesser dose required to reach efficacy should be used [33]. Minimum dosing interval: 4 hours
IV infusion: Use pre-filled vial 1000 mg/100 mL, no dilution is necessitated [34]. Infuse dose intravenously over 15 minutes [35]. Where doses less than a full vial are necessitated (i.e. weight < 50 kg), draw up the exact dose from the vial for administration via a syringe attachment or if a syringe attachment is not applicable (e.g. volume above 50 mL) withdraw and dump the amount not necessitated from the vial before administration [36, 37].
Concentration: 10 mg/mL [38].
Populations: Children: Paracetamol-caffeine is not recommended for children under the age of 12 years [39].
Renal Impairment: Patients who have been diagnosed with liver or kidney impairment must need medical counsel before receiving this medicine. The challenges related to the use of paracetamol and caffeine products in patients with renal impairment are initially an outcome of the paracetamol content of the medicine [40].
Hepatic Impairment: Patients who have been diagnosed with liver or kidney impairment must need medical counsel before receiving this medicine [41, 42].
Pregnancy: High amount of data on pregnant women indicate neither malformative, nor feto/neonatal toxicity. Epidemiological surveys on neurodevelopment in children exposed to paracetamol in utero reveal inconclusive sequences [43, 44]. Paracetamol-caffeine is not recommended for use during pregnancy owing to the probable increased risk of spontaneous abortion associated with caffeine drink [45, 46].
Breastfeeding: Paracetamol is excreted in breast milk but not in a clinically important amount. No negative outcomes on infants have been reported. Paracetamol perhaps used by breastfeeding women as long as the recommended dosage is not exceeded [47].
ADR: Hepatobiliary disorders [48]; Cardiac disorders [49]; gastrointestinal disorders [50]; Psychiatric disorders [51]; Blood and lymphatic system disorders [52]; Metabolism and nutrition disorders [53]. Hepatotoxicity of paracetamol and related fatalities: In liver microsomes, a least percentage of paracetamol (5-10%) is transformed by CYP P450 isoforms (CYP2E1, CYP2A6) into a reactive metabolite, NAPQI that is initially related to paracetamol hepatotoxicity. About 2% of paracetamol is excreted in urine unchanged [54]
Pharmacological Properties
Pharmacodynamic properties: Pharmacotherapeutic group: Other analgesics and antipyretics, anilides. The dearth of peripheral prostaglandin suppression gives substantial pharmacological properties such as the maintenance of the protective PGs within the GIT. Paracetamol is, therefore, specifically favourable for: patients with a history of damage or patients receiving coincident medicines, where peripheral PG suppression would be unwanted (such as, for instances, those with a history of GI bleeding or the elderly) [55-57]. Caffeine acts as an analgesic adjuvant which accelerates the efficacy of paracetamol [58].
Pharmacokinetic properties
Absorption: After oral administration paracetamol is hastily and nearly comprehensively absorbed. Peak plasma concentrations are achieved after 30 minutes to 2 hours [59].
Distribution: Paracetamol is distributed hastily throughout all tissues. Concentrations are analogous in blood, saliva and plasma [60]. The volume of distribution of paracetamol is comparatively 1 L/kg bodyweight. At therapeutic doses protein binding is insignificant [61].
Metabolism: In adults paracetamol is conjugated in the liver with glucuronic acid (~60%), sulphate (~35%) conjugates. The subsequent route is hastily saturated at doses greater than the therapeutic dose. A minor route, catalyzed by the CYP P450, sequences in the formation of an intermediate reagent (N-acetyl-p-benzoquinoneimine) which under normal circumstances of use is hastily detoxified by glutathione and eliminated in the urine, after conjugation with cysteine (~3%) and mercaptopuric acid [62, 63]. In neonates and children <12 years sulphate conjugation is the chief elimination route and glucuronidation is lesser than in adults. Total elimination in children is analogous to that in adults, owing to an increased capacity for sulphate conjugation [64].

Figure 3: mechanism of paracetamol metabolism
Elimination: Elimination of paracetamol is indispensably through the urine. 90% of the ingested dose is eliminated via the kidneys within 24 hours, substantially as the glucuronide (60 to 80%) and the sulphate (20 to 30%) conjugates. Less than 5% is eliminated in unchanged form. The elimination half-life is about 2 hours [65]. In cases of renal or hepatic inadequacy, after toxicity, and in neonates the elimination half-life of paracetamol is holding pattern. The maximum outcome is same with plasma accumulations. For elderly patients, the capacity for conjugation is not changed [66].
Contraindications: This product is contraindicated in patients with a former history of hypersensitivity to paracetamol (caffeine or excipients).  For patients with severe hepatocellular inadequacy, hepatic failure or decompensated active liver damage not used [67, 68].
For patients with severe hepatocellular inadequacy, hepatic failure or decompensated active liver damage not used [67, 68].
Antidote: N-Acetylcysteine: Acetylcysteine AKA N-acetylcysteine prevents the hepatic damage, initially by renewing hepatic glutathione. It is considered to provide cysteine for the glutathione generation and conceivably to figure an adduct directly with the toxic metabolite of acetaminophen and N-acetyl-p-benzoquinoneimine and to thus inhibit its covalent bonding to the hepatic proteins [69, 70]
Conclusion
Paracetamol is a common analgesic that acts as a competitive inhibitor of COX enzymes. It is metabolised in the liver through several pathways, involving glucuronidation and sulfation, to accelerate its excretion from the body. Paracetamol has chiefly anti-pyretic (decreasing the levels of PGs in the hypothalamus) and analgesic properties; it does not interfere with COX 2 and does not affect the different constituents of inflammation (swelling and redness). Probenecide prevents the binding of paracetamol to glucuronic acid decreasing paracetamol clearance by a factor of about 2. If probenecide is taken coincidently the paracetamol dose should be decreased.
Abbreviations
COX-1: cyclooxygenase type 1; COX-2: Cyclooxygenase type 2; CYP450: cytochrome P450; DI: Drug interaction; PG: Prostaglandin; NSAIDs: Non-steroidal anti-inflammatory drugs; NAPQI: N‐acetyl‐p‐benzoquinone imine
Acknowledgments
The author would be grateful to anonymous reviewers for the comments that increase the quality of this manuscript.
Data Sources: Sources searched include Google Scholar, Research Gate, PubMed, NCBI, NDSS, PMID, PMCID, and Cochrane database. Search terms included: paracetamol medication properties
Funding
None












Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org