Clinical Case Reports and Clinical Study
OPEN ACCESS | Volume 13 - Issue 1 - 2026
ISSN No: 2766-8614 | Journal DOI: 10.61148/2766-8614/JCCRCS


Maria Virginia Pereira Gomide Freitas1, Maria Goretti Moreira Guimarães Penido1,2*
1Unidade de Nefrologia Pediátrica, Departamento de Pediatria, Hospital das Clínicas, Faculdade de Medicina da Universidade Federal de Minas Gerais, Brasil.
2Unidade de Nefrologia Pediátrica - Centro de Nefrologia da Santa Casa de Belo Horizonte, Minas Gerais, Brazil
*Corresponding Author: Maria Goretti Moreira Guimarães Penido, Centro de Nefrologia da Santa Casa de Belo Horizonte R. Piauí, 420 - Santa Efigênia, Belo Horizonte - MG, 30150-320, Brazil.
Received: February 11, 2023
Accepted: February 15, 2023
Published: February 20, 2023
Citation: Maria Virginia Pereira Gomide Freitas, Maria Goretti Moreira Guimarães Penido (2023) “Nephropathy Due to Immunoglobulin A Deposits: Advances in Pathogenesis”, Clinical Case Reports and Clinical Study, 1(9); DOI: http;//doi.org/02.2023/1.162.
Copyright: © 2023 Maria Goretti Moreira Guimarães Penido. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly Cited.
IgA nephropathy is considered a distinct form of primary glomerulonephritis. In most cases, the initial clinical presentation is isolated, intermittent or persistent, macro or microscopic hematuria of glomerular origin. However, arterial hypertension and proteinuria may appear in varying degrees. In the 60s it was considered a benign disease with a favorable prognosis, however, prospective studies with long years of follow-up have shown disease progression to complete loss of renal function in pediatric and adult patients.
Evidence demonstrates that IgA nephropathy, is an autoimmune disease wherein the kidneys are damaged as innocent bystanders due to deposition of IgA1-IgG immune complexes from the circulation. Patients with IgA nephropathy have elevated circulating levels of IgA1 with some O-glycans deficient in galactose (galactose-deficient IgA1) and these IgA1 glycoforms are recognized as autoantigens by unique IgG autoantibodies. These circulating immune complexes deposit in the glomeruli and activate mesangial cells that respond with mesangial hypercellularity, apoptosis, oxidative stress, complement activation, mesangial matrix expansion, damage to podocytes and proximal tubule epithelial cells, increased glomerular permeability and fibrosis in the glomerular and interstitial compartments. Clinically, these renal lesions will cause hypertension, proteinuria, hematuria, and reduced renal function.
The immunopathogenesis of the disease is still not fully understood and further studies are needed. With a better understanding of immunopathogenesis of IgAN it will be possible an earlier diagnosis and more appropriate treatment.
1. Introduction
Nephropathy due to immunoglobulin A deposits (IgAN) is the most common primary glomerulopathy worldwide and remains an important cause of chronic kidney disease (CKD) and end-stage kidney disease (ESKD).1,2 Also known as Berger's Disease, it has been considered a clinical-pathological entity since its description by Berger and Hinglais in 1968.3 Although it is common at all ages, the frequency is higher in the second and third decades of life.4,5
The incidence is 2–10 per 100,000 persons per years5,6-11 with a peak during the second and third decades of life.12,13 In European countries the incidence ranges from 15 to 40 new cases per million population per year (pmpy). In Japan, the calculated incidence in children was 4.5 cases/pmpy.8 The male to female ratio is 1:1 in Asia14, 2 –3:1 in North America4,5,15 and Europe.16
The prevalence differs geographically and between ethnic/racial groups, being highest in people of East Asian descent, followed by Caucasians. It is rare in individuals of sub-Saharan African ancestry.6-8 IgAN corresponds to 20-40% of glomerulonephritis in countries such as Japan, Singapore, France, Italy, New Zealand and Australia, and two to 10% in the United States, Canada and England.17 These differences result from specific research programs for the diagnosis of glomerulonephritis, racial, genetic and environmental factors.4,18 In Japan, all children between six and 15 years old are assessed annually and referred for further investigation if necessary.17,19
Some relatives with familial IgAN have been described, although most cases of the disease appear to be sporadic.20
Primary IgAN is an immune complex-mediated glomerulopathy defined histologically by the presence of immunoglobulin A (IgA) deposits accompanied by a variety of histological lesions.21 However, its pathogenesis and the source of the IgA deposits are not completely understood.22,23 Patients diagnosed with IgAN do not share a single pathogenetic mechanism and mesangial deposition may be the final expression of a common pathway for more than one type of IgA immune system abnormality.22-30 Although primary glomerulopathy receives more attention, many diseases are associated with glomerular deposits of IgA, being the Henoch-Schönlein Purpura (HSP) the most common, and may represent the systemic form of primary IgAN30. Isolated, intermittent or persistent, macro or microscopic hematuria of glomerular origin is the most frequent initial clinical presentation, however, arterial hypertension and proteinuria can appear in varying degrees.
IgAN was considered a benign disease but is currently recognized as the main cause of renal failure in patients with primary glomerular disease undergoing a renal replacement program. Prospective studies with long years of follow-up have shown progression to CKD in children and adults.31-41 Progression to renal failure has been demonstrated in 20 to 50% of adult patients after 10 to 20 years of follow-up.35 Also in pediatric patients, studies in France,37 Japan,31 Sweden,34 Germany,36 United States32-33 and Italy39 demonstrated progression to progressive loss of renal function. Yoshikawa et al., in 1989, found that 5% of children with IgAN progressed to CKD within 5 years after disease onset, 6% within 10 years and 11% within 15 years of disease.42 Hogg et al., in 1994, carried out a multicenter study involving 13 American centers of pediatric nephrology. The authors evaluated 218 children and adolescents with IgAN. Among these patients, 80 were followed carefully for four years and 12 patients (15%) evolved to CKD.29
Some patients enter in sustained clinical remission (normal renal function, urine without hematuria and proteinuria, and normal blood pressure). However, renal biopsies usually reveal persistent glomerular deposits of IgA.43,44 In contrast, patients with persistent urinary changes have shown progression of glomerular damage.44,45 Patients who progressed to dialysis or death had 3 risk factors at the time of renal biopsy: proteinuria ≥ 1 g/day, sustained arterial hypertension, and severity of renal involvement based on the revised Oxford classification of IgAN.46-48
Histopathologically, IgAN is characterized by the presence and predominance of IgA deposits in the glomerular mesangium in a diffuse manner, accompanied by varying degrees of focal or diffuse proliferation, in the absence of any other systemic disease. In the last decade there has been a substantial expansion of knowledge about various aspects of the pathogenesis of IgAN.21-28
Renal biopsy is required for the diagnosis of IgAN. The presence of IgA as the dominant or codominant immunoglobulin in the immunological deposits in the mesangial areas of the glomeruli defines the disease and is demonstrated by immunofluorescence.49 IgA is the only immunoglobulin deposited in 15–40% of cases. In the remaining cases, IgG, IgM or both are present.50 Glomerular IgA is exclusively of the IgA1 subclass, which has specific characteristics that greatly contribute to the pathogenesis of the disease. Other immune proteins can be detected by immunofluorescence microscopy. Complement component C3 is co-localized with IgA in more than 90% of biopsies with IgAN.51 C3, C4, C4d,28 properdin, terminal complement complex (C5b-C9),52 and mannose-binding lectin53 are frequently detected. C1q is typically absent.54-57 These features support the involvement of alternative pathways and complement activation lectin in the pathogenesis of IgAN.
2. Pathogenesis
IgA is the most abundant antibody in secretions, representing 10-15% of all circulating immunoglobulins. In humans, the first B cells with IgA appear in the 11th week of life and adult levels are only reached at puberty.58 It is the immunoglobulin that contributes to the immunity of the excretory system and can be found in the body in two compartments: IgA that circulates freely in the plasma and secretory IgA that is secreted in the mucous membranes. There are two subclasses of IgA: IgA1 and IgA2, different due to the number of amino acids. This structural difference explains IgA2 resistance to bacterial proteases and also why this subtype is more frequent in the mucous membranes.58 IgA exists in both monomeric and polymeric form. In healthy individuals 95% of circulating IgA is in monomeric form and is produced by circulating lymphocytes and plasma cells in the spleen and bone marrow. In contrast, the IgA produced by plasma cells of the respiratory and intestinal tracts is in the dimeric form and is formed by two monomeric IgA linked by a chain called J.59 For these dimers to be secreted on the mucosal surface, they must be bound to a specific receptor (RR), the polymeric IgA receptor, present at the basal pole of the epithelial cell. After binding to the receptor, the IgA/RR dimer complex is internalized and transported to the luminal pole of the epithelial cell. In this location, the two IgA linked by the J chain are released on the mucosal surface, preserving the RR fragment, known as the secretory component.59,60 In healthy individuals, mucosal IgA does not reach circulation.59
Fifty to 70% of patients with IgAN have increased serum levels of IgA and deposits in the renal mesangium are mainly of the IgA1 subclass, with their nephritogenicity dependent on their polymeric nature, molecular structure and ionic charge.42 Studies show that cultures of peripheral blood lymphocytes from patients with IgAN produce more IgA than normal individuals and that this increase in IgA production remains stable in patients with persistent hematuria and that it is reduced or normal in patients in clinical remission. The regulation of IgA production is highly dependent on T cells and increased production of this immunoglobulin indicates alteration in T cell function.61
Advances have been made in understanding the pathogenesis of IgAN. The available evidences suggest that there are different mechanisms for IgA deposition in the renal mesangium.58 These advances have shown that it is very likely that there is not just one type of IgAN with a simplified pathogenetic mechanism. As aforementioned, this IgA is restricted to the IgA1 subclass42 and has less galactose in its O-glycans than does circulating IgA1in healthy individuals (galactose-deficient IgA1: Gd-IgA1).62,63 The immune proteins in the glomeruli of patients with IgAN can include complement C3; IgG, IgM, or both.2,51.
Knoppova et al., have postulated that IgAN is an autoimmune disease with a multi-hit mechanism: 1) patients with IgAN produce large amounts of Gd-IgA1; 2) these Gd-IgA1 are recognized as autoantigens by IgG autoantibodies; 3) there are formation of immune complexes in the blood; 4) deposit in the glomeruli and activate the mesangial cells, inducing mesangial lesions.57 Thus, this hypothesis also suggests that the development of kidney disease must occur in four main stages: 1) deposit of IgA in the mesangium; 2) generation of mesangial injury mediated by the interaction of IgA1 complexes with specific receptors or through complement activation; 3) progression of the mesangial lesion to renal failure39. In most patients, it is not possible to demonstrate the presence of the secretory component in the mesangium, however, it is considered that IgA1 originates from the plasma cells of the bone marrow.57,58
This sequence is in agreement with observations that serum levels of Gd-IgA1 (autoantigen) and the corresponding antibody correlate with disease severity and progression.50,64-66 Glomerular co-deposits of IgG are of the IgG1 and IgG3 subclasses67 as are IgG autoantibodies in the circulation64 and their levels correlate with those of the autoantigen, Gd-IgA1,68 predicting disease progression and recurrence after kidney transplantation.50,66,69-71
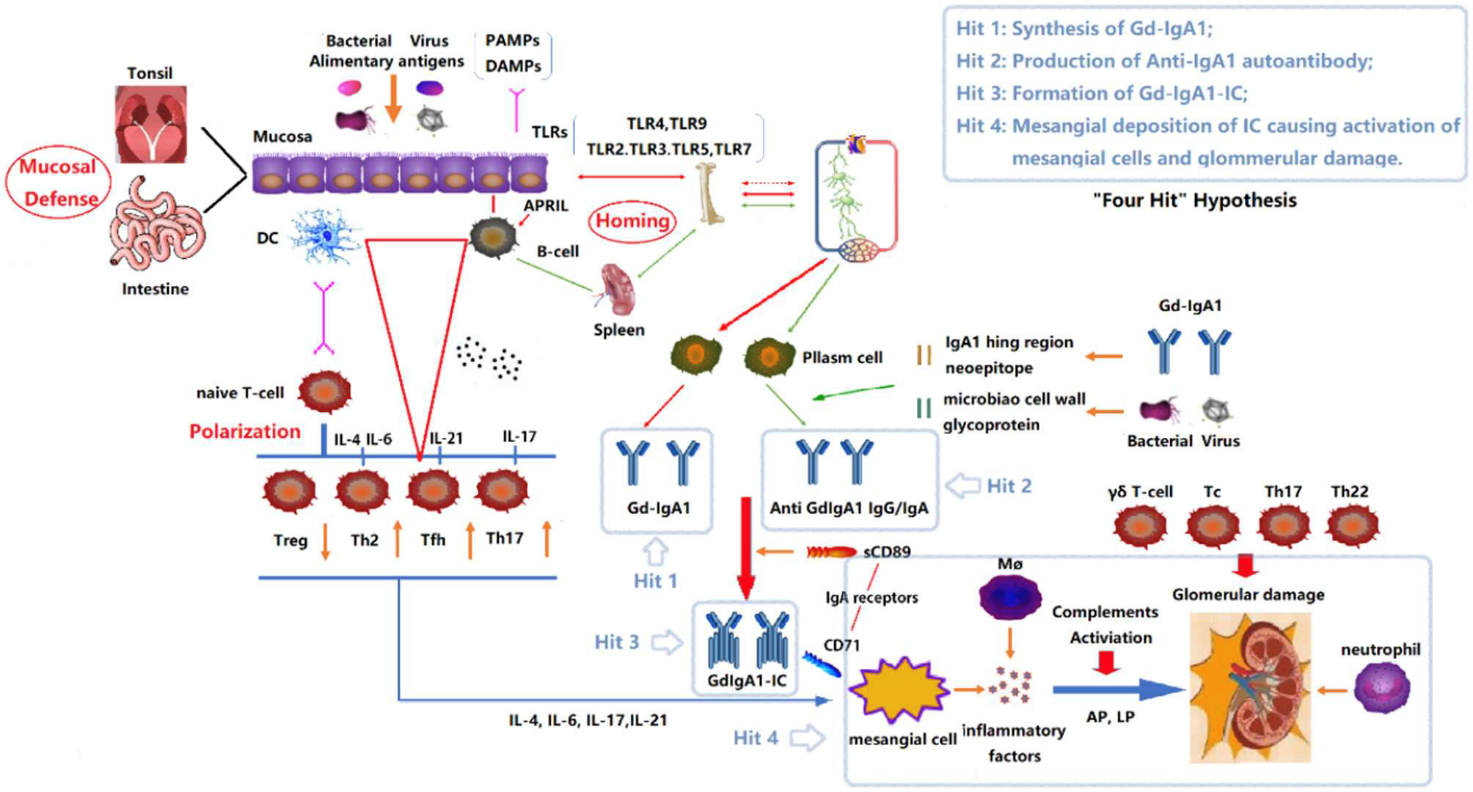
FIGURE 1 “Four Hits” Hypothesis of IgA Nephropathy. In individuals with a genetic predisposition to IgA nephropathy, infection, or other events destroy the mucosal barrier defense function. Chronic stimulation such as pathogenic microbial or alimentary antigens are taken up by antigen-presenting cells, thereby activating B cells, and differentiating into plasma cell secreted IgA in T-cell-dependent or non-dependent manners. Due to the abnormal regulation of mucosal-bone marrow axis, the mis-expression of homing receptors on the surface of B/plasm cells leads to increased synthesis of poorly glycosylated IgA1 (Gd-IgA1) and self-aggregation to form aggregated Gd-IgA1 (Hit 1). The aberrant exposure of GalNAc of Gd-IgA1 as antigen stimulates B cells to differentiate into plasma cells and synthesizes anti-Gd-IgA1 autoantibodies (Hit 2). The Gd-IgA1 immune complex (Gd-IgA1-IC) is formed by anti-Gd-IgA1 autoantibodies binding to Gd-IgA1 along with soluble sCD89 (Hit 3). Macromolecular Gd-IgA1-IC binds to the IgA receptor (CD71) expressed on mesangial cells and deposits on the glomerular mesangium. Subsequently, the mesangial cells release various inflammatory factors (cytokines, chemokines, growth factors, etc.) under the stimulation of ICs, attracting and recruiting multiple subsets of T cells, macrophages, neutrophils infiltration, and activating the lectin pathway and alternative pathway of the complement system. Synergistic effects lead to glomerular injuries such as mesangial hyperplasia, matrix expansion, and interstitial fibrosis (Hit 4). DC, Dendritic cell; TLRs, Toll like receptors; APRIL, a proliferation-inducing ligand; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; Gd-IgA1, poorly glycosylated IgA1/Galactose-deficient
IgA1; AP, The alternative pathway; LP, The lectin pathway.76
2.1 Mesangial IgA deposits
Immunoglobulin A in healthy individuals is folded in the region between amino acids 223 and 240 and contains serine and threonine which, under normal conditions, are glycosylated by the union of N-acetylgalactosamine (GAlNAc). This serine or threonine / GalNAc complex incorporates galactose through the action of the enzyme beta-1,3 galactosyltransferase and forms the disaccharide Gal-GalNAc. This disaccharide, through the action of the alpha-2,3 sialotransferase enzyme, can incorporate one or two sialic acid units.57,72-73 Abnormalities in these incorporations can prevent effective glycosylation of IgA and produce hypo glycosylation. Despite efforts, the main cause of hypo glycosylation remains unclear. Studies have shown the autoimmune characteristics of IgAN.57,76 As already mentioned, this autoimmune character of the disease is explained by a multi-hit model of pathogenesis. There is an overproduction of aberrantly glycosylated IgA1, deficient in galactose in some O-glycans, by IgA1-secreting cells leads to elevated levels of circulating galactose-deficient IgA1. There is production of autoantibodies that bind to galactose-deficient IgA1 molecules in the hinge region, resulting in the formation of nephritogenic immune complexes. These complexes can deposit in the kidneys, activating mesangial cells and initiating glomerular injury. Thus, galactose-deficient IgA1 is the key to the onset of the disease.42,44,57,73-76
The following sequence of proposed pathogenetic mechanisms is:
1) Aberrantly glycosylated IgA1 has an increased ability to aggregate other molecules of abnormally glycosylated IgA1 forming polymers, has increased affinity for different components of the mesangial matrix and can induce an immune response forming circulating immune complexes that are nephritogenic.43-45,57 Thus, an increased amounts of circulatory Gd-GdIgA1 are recognized as an autoantigen by autoantibodies (mostly of the IgG subclass);44
2) The polymers formed by the self-aggregation of aberrantly IgA1 interact with the alpha Fc receptor (CD89) which is expressed on the surface of lymphoid and mononuclear cells. This receptor represents a transmembrane protein that binds to IgA1 with low affinity. Patients with IgAN have reduced membranous expression of this receptor, although its intracellular synthesis is normal. Monteiro et al., demonstrated the presence of this receptor linked to IgA in the form of circulating complexes (CD89-IgA) in the blood of patients with IgAN but absent in healthy ones.60 Studies showing the anomalies observed in the CD89 of patients with IgAN and evidence in mice suggest that the fixation of abnormal IgA on CD89 would provide the release of the extracellular portion of the receptor and decrease the membranous CD89.60,61 This extracellular part of the CD89, once in the circulation, would bind to the IgAs forming the extended soluble IgA-CD89 complexes and, as a result, there would be a decrease in the number of CD89 on the surface of the cells.57,60,61 These extended soluble IgA-CD89 complexes, or also called IgA RR-alpha Fc complexes (FcαRI), are not recognized by the liver and there is a decrease in the clearance of polymeric IgAs, an increase in their serum concentration and their interaction with CD89. These complexes are deposited in the mesangium through binding to receptors on the surface of mesangial cells.22,60,61,77 The detection of circulating complexes containing IgA and soluble FcαRI (CD89) in the serum of patients with IgAN increases the possibility that these complexes are strongly involved in the pathogenesis of the disease.
3) Finally, the region where IgA1 is folded and with aberrant glycosylation is antigenic and induces the production of circulating IgG antibodies, originating polymeric IgA1-IgG immune complexes. The aberrant IgA1 can also interact with other circulating proteins such as fibronectin, laminin and collagen.12 The end result of these processes is the formation of aggregated macromolecular and immune complexes that persist in the circulation, not recognized by hepatic receptors and not degraded, and then deposited in the renal mesangium due to affinity with extracellular matrix proteins or through interaction with specific mesangial receptors.44,60,61,77
It has been described that renal biopsies from patients with IgAN have positive staining for mannose-binding lectin (MBL), L-ficolin and C4d48. This positivity indicates that there was complement activation through the lecithin pathway and studies have associated this with worse prognosis.78 The reason for the presence of MBL in some renal biopsies of these patients is not known and the relationship between these deposits and their circulating levels has not been proven. However, it has been shown that only polymeric IgA1 is able to interact with MBL and there is an association between MBL polymorphisms and the clinical evolution of NigA, but not with the prognosis.58,79-81
It is important to note that high levels of aberrantly glycosylated circulating IgA1 alone do not induce glomerular injury.82 Additional factors are known to be involved in the development of IgAN, namely genetic factors, mucosal immunity and the antigenicity of Gd-IgA1 related to autoantibody production.83,84
2.2 Interaction between IgA1 complexes and mesangial receptors
Although many proteins were suggested as possible candidates for mesangial IgA receptors, none of them were found in the mesangium. However, there are five well-known IgA receptors: FcaR1 (CD89), asialoglycoprotein receptor (ASGPR), polymeric Ig receptor (pIgR), transferrin receptor (TfR: CD71) and Fc a/m receptor. Some of these receptors are involved in removing IgA from the circulation, such as ASGPR, CD89.85 TfR binds only polymeric and not monomeric IgA1, and in contrast to FcαRI, this IgA receptor is only weakly expressed in mesangial cells.86 Interestingly, in patients with IgAN, TfR is strongly expressed in the mesangium and correlates with disease severity.86 Probably, the TfR/CD71 receptor participates in the selective deposition of IgA1 complexes because it has an overexpression in the mesangium of patients with IgAN.86,87 Similar observations have been found in patients with glomerulonephritis of Henoch-Schoenlein purpura with mesangial IgA deposition and in patients with lupus who have mesangial IgA deposition.86 Studies have indicated that abnormally glycosylated IgA1 and IgA1 complexes may favor interaction with TfR as observed in normal human mesangium cell cultures.88 Furthermore, IgA1 polymers can induce TfR expression, cytokine release and cell proliferation, which could partly explain the observed tissue injury and recurrence of deposits after kidney transplantation.88 Additional receptors able to bind IgA1 were identified in human mesangial cells: integrin a1/b1 and a2/b1 and b1,4-galactosyltransferase 1.89,90 Expression of the latter receptor in glomeruli is increased in IgAN, indicating that b1,4-galactosyltransferase 1 may play a role in IgA clearance and in the initial response to IgA deposition.89,91 This observation on the role of b1,4-galactosyltransferase was reported earlier for binding of IgA1 to various cells.92
Another described receptor for IgA and IgM is the Fcα/µR.93 This receptor is probably regulated by IL-1, implying another mechanism for mesangial IgA deposition. However, this receptor has not been found in cultured human mesangial cells,94 but it still cannot be excluded in some cases of IgAN with IgA and IgM deposits and with severe inflammatory process.
2.3 Complement Activation and immune Modulation
There are three complement activation pathways: classical pathway (CP), the alternative pathway (AP) and the lectin pathway (LP). Each of these pathways has a respective activation mechanism. Glomerular co-deposition of IgA and C3 complement components are frequent in IgAN.95 Thus, activation of the complement pathway is thought to play an important role in glomerular injury in this disease.96 Co-deposition of C3b, factor P, properdin and factor H (FH) is observed in almost 100%, 75-100% and 30-90% of cases, respectively.96,97. Data suggest that alternative complement and lectin pathways are involved in the pathophysiology of IgAN.98 Have been found patients with IgAN that showed deposition of: a) MBL-associated C4d, b) mannose-binding lectin (MBL), c) Ficolin and serine protease deposition, d) membrane attack complex (MAC), and associations of MBL and L-ficolin deposition.53,76. The alternative complement pathway is considered the most important pathway in the pathogenesis of IgAN. Reports suggest a relationship between complement activity and IgA1 glycosylation abnormalities. Studies have shown that MBL binds to IgG and IgM N-glycans99,100. Therefore, N-glycosylation of polymeric IgA could be associated with MBL binding and activation of the lectin pathway. N-glycans of the heavily glycosylated secretory component may also contribute to complement activation, as seen in IgAN.101 More studies are needed to determine which N-glycan components and structures are involved in lectin pathway activation and whether IgA1 N- or Oglycosylation are involved in alternative complement activation pathways.102
It has been suggested that immunological factors are involved in all aspects of IgAN development and may play an important role. It is believed that a wide variety of immune cells (dendritic cells, macrophages, subsets of T lymphocytes and B lymphocytes, etc.) and molecules (IgA receptors, Toll-like receptors, complements, etc), that are part of innate and adaptive immunity, are involved in the pathogenesis of IgAN. The role of immunity of tonsils or intestinal mucosa is supported by increasing evidence.76
Physiologically, T cells activated by mucosal antigens (bacterial or viral products, food antigens, etc.) trigger naive B cells to switch from class IgM to IgA in a dependent T cell in Peyer's patches and tonsils. Activated B cells migrate to regional lymph nodes and differentiate into IgA-secreting plasma cells. IgA dimers move through the mucosal epithelium, become SIgAs, which act in defense against pathogens. The continuous exogenous antigenic stimulus, abnormal immunity of the mucosal response, and the incorrect expression of homing receptors in the surface of IgAl-secreting B/plasma cells can result in aberrantly glycosylated IgA1. Soluble CD89/Gd-IgA1-IgG complexes are deposited in the mesangium by binding to their receptors and continuous release of cytokines and growth factors into the mesangium, with inflammatory damage, matrix accumulation, and glomerulosclerosis. Recruitment of T lymphocytes leads to renal tubular interstitial damage and fibrosis.76,103
As for the role of Tc cells in the pathogenesis of IgAN, it was mentioned that high expression of CX3CR1 (chemokine receptor - CX3CR1) is present in the CD8+ Tc peripheral blood of patients with IgAN. This increases lymphocytes that move across the glomerular endothelium causing destruction of the glomerular capillary wall and, consequently, hematuria.104,105
3.Induction Of Inflammatory Mediators And Facilitation Of Kidney Disease Progression
IgAN is a disease characterized by the deposition of immune complexes containing Gd-IgA1 and IgG autoantibodies specific for Gd-IgA1,83,106,1097 and is an autoimmune disease. These glomerular immunodeposits usually contain the C3 fraction of the complement. It is believed that these glomerular immunodeposits originate from the circulation .108 In vitro experiments with primary human mesangial cells have shown that immune complexes containing IgA1 in the serum of patients with IgAN can activate mesangial cells.86,109-111 In contrast, uncomplexed free Gd-IgA1 did not stimulate mesangial cell proliferation.110-112 It is not yet known what type of mesangial cell receptors are involved by the pathogenic complexes, but mesangial cells express several receptors that can bind IgA1. In general, IgA complexes can induce immunosuppressive or pro-inflammatory responses.
Although IgA deposition and the onset of glomerular inflammation is a process specific to IgAN, the inflammatory processes are similar to those seen in other glomerular diseases and include alterations in mesangial cell proliferation/survival in the contractile or secretory state, resulting in cell acquisition mesangial of a pro-inflammatory and pro-fibrotic phenotype.61 In addition, mesangial cells produce platelet-derived growth factor beta chain (PDGFB) which induces mesangial proliferation contributing to progressive kidney disease.22-24, 57,58,60,61
As already mentioned, the central event for glomerular injury is the binding of IgA immune complexes containing poorly galactosylated IgA1 O-glycoforms with mesangial cells and its consequent activation. This binding results in proliferation and release of pro-inflammatory and pro-fibrotic mediators.93,113,114 These mediators cause podocyte injury, scarring 115 and proximal tubular epithelial cell (PTEC) activation, which drives tubulointerstitial scarring.115,116 Concomitantly, there is local and systemic activation of complement C3. After C3 activation, C5b-9 is generated, whose concentrations can also activate mesangial cells to produce inflammatory mediators and matrix proteins.2,117 Activated mesangial cells secrete components of the extracellular matrix, increase the expression of inducible nitric oxide synthase, and release various mediators of renal injury: angiotensin II, aldosterone, pro-inflammatory and pro-fibrotic cytokines, and growth factors.109,118,119 These events, if prolonged, cause mesangial hypercellularity, apoptosis, oxidative stress, complement activation, mesangial matrix expansion, damage to podocytes and proximal tubule epithelial cells, increased glomerular permeability and fibrosis in the glomerular and interstitial compartments.109,119,120 Clinically, these renal lesions will cause hypertension, proteinuria, hematuria, and reduced renal clearance.109,119
The renin-angiotensin system (RAS) has been implicated in the development of progressive glomerulosclerosis in diabetic and non-diabetic nephropathy.121 Angiotensin II (AII) plays a key role in glomerulosclerosis by inducing TGF-β expression in mesangial cells. TGF-b stimulates extracellular matrix protein synthesis, increases matrix protein receptor levels and alters the balance of proteases/protease inhibitors and therefore inhibits matrix degradation.121 TGF-b is involved in the development of glomerular and tubular fibrosis. Mesangial cells from patients with IgAN produce TGF-β and there is also increased gene expression of TGF-β in circulating CD4+ T cells in these patients.121
Lai et al. demonstrated that pIgA1 from patients with IgAN directly stimulated the RAS in mesangial cells which activates the synthesis of TGF-b causing formation and accumulation of extracellular matrix, trans differentiation of myoblasts of the tubular epithelium and renal fibrosis. In vitro, RAS blockade with angiotensin-converting enzyme inhibitor (ACE) or angiotensin receptor blocker (ARB) suppressed TGF-b synthesis, preventing the development of renal fibrosis. The upregulation of renin and AII by pIgA1 in mesangial cells suggests that early and aggressive blockade of the RAS is essential for preventing progression of renal injury.
Wyatt et al., suggested that alterations in the complement system, together with hereditary complement deficiencies could be important for the clinical expression of IgAN in individuals with susceptibility to it. It is known that activation of the complement system can occur in IgAN with glomerular deposits of C3, both in adults and in children, and that serum concentrations of this C3 can be normal or altered.15 Likewise, Masashi et al., demonstrated that IgA levels strongly correlate with the percentage of CD4+ and CD5+ monoclonal antibodies in the peripheral blood of these same patients. There are still reports of an association between the HLA system and IgA nephropathy, despite conflicting results.122
Studies have shown that transmembrane FcαRI activation and interaction with IgA-IC aggravates IgAN through induction of cytokine and chemokine cascade and leukocyte infiltration.123 Another factor that contributes to the evolution of the disease to ESKD is the overexpression of TfR by mesangial cells that facilitates the mesangial deposition of IgA1-IC.85-88,93 This interaction triggers an inflammatory response promoting the release of pro-inflammatory cytokines such as IL-1, IL-6 and TNF-α, with consequent cell proliferation and fibrosis.85-88,93
The severity of IgAN is often associated with leukocyte infiltration, and disease progression is associated with the presence of monocytes, macrophages, and T cells.125-126 Mesangial proliferation and the presence of glomerulosclerosis are related to increased IgA uptake by blood phagocytes of patients with IgAN.85 FcαRI-deficient endocytosis and increased supply of IgA has been demonstrated in IgAN126.
Finally, evidence demonstrates the importance of aberrantly O-glycosylated IgA1 and the corresponding IgG autoantibodies in the formation of nephritogenic immune complexes. However, further studies are needed to fully understand the pathogenesis of IgAN.
4.Genetic Factors
The identification of several members of the same family with IgAN suggested a genetic predisposition.128 Genome-wide association studies (GWAS) favored the discovery of other variants in the human genome that influence the pathogenesis of the disease. To date, 18 susceptibility segments (loci) of the genome have been identified that modulate IgAN risk and blood levels of Gd-IgA1 are an inherited trait.129-131
Approximately 75% of patients with IgAN and 30-40% of their first-degree relatives have serum Gd-IgA1 levels above the 90th percentile when compared to healthy persons.132 GWAS identified variants in 2 genes encoding enzymes important for IgA1 O-glycosylation and these variants were associated with higher serum levels of Gd-IgA1.133-134 The C1GALT1 gene encodes the human core enzyme 1 β1–3-galactosyltransferase (C1GALT1), required for the addition of galactose to N-acetylgalactosamine on the glycans of the hinge region of IgA1. The C1GALT1C1 gene encodes a molecular chaperone, COSMC, which stabilizes the activity of the C1GALT1 enzyme.
The activity of these enzymes is reduced when there are variants of both genes. Consequently, a smaller amount of galactose would be bound to N-acetylgalactosamine in the glycans of the hinge region of IgA1, increasing the synthesis of Gd-IgA1. Studies have confirmed that messenger RNA levels for these genes determine the rate of Gd-IgA1 secretion in immortalized IgA1-producing cells.133 These findings indicate a genetic influence in the regulation of the synthesis of Gd-IgA1, the autoantigen of IgAN.
GWAS analyzes also showed that variants of several genes that influence immune response or antigen presentation are associated with IgAN. These include 3 loci in the major histocompatibility complex and the CFH and CFHR genes that encode complement factor H (CFH) and CFH-related proteins (CFHR), respectively, which are components of the alternative complement pathway.44,133,134 The risk of having IgAN is 30% lower for patients with the variant in both genes.135 Histocompatibility complex genes that encode human leukocyte antigens (HLA), including HLA-B, DRB1, DQA, and DQB, and genes at non-HLA loci that encode variants of angiotensinogen, angiotensin converting enzyme (ACE), and angiotensin type 1 receptor II are within the other genetic loci associated with IgAN.135,136.The frequency distribution of the 18 IgAN-associated risk variants correlated with ethnic differences in disease prevalence, highest in Chinese, medium in Caucasians, and lowest in Black Africans.137
The role of the megsin gene and other genes in patients with IgAN has been investigated.138 Megsin (SERPINB 7) is a gene expressed predominantly in the mesangium and belongs to the serpin (serine proteinase inhibitor) superfamily. Megsin was cloned in 1998 and has been implicated as a gene associated with or contributing to various mesangial lesions.139 Immunohistochemical and in situ hybridization studies have shown that it is up-regulated in IgAN when compared to normal tissue and other forms of glomerulonephritis.139 Megsin upregulation coincides with mesangial proliferation and extracellular matrix expansion. The authors showed that the 2093C and 2180T alleles were transmitted more frequently from heterozygous parents to affected patients than would be statistically expected. The results indicate that, in the studied population, genetic variations or megsin variations confer susceptibility to IgAN.139
Thus, the disease presents a complex multifactorial genetic distribution pattern with polygenic and environmental causal factors. Therefore, IgAN does not follow a clear pattern of inheritance, but there is a familial aggregation of the disease. A number of polymorphic genes with small additive effects encode disease susceptibility. Environmental factors acting in genetically susceptible individuals can trigger the process leading to clinical expression of the disease.140
5. Conclusions
Despite the great advances in the understanding of IgAN in the last decades, many doubts still persist. It is known that the prognosis of this disease is not always benign and studies try to find minor clinical factors that could indicate a bad evolution of the disease and that would guide the appropriate conduct. The identification of clinical and/or laboratory parameters that allow us to predict which patients will evolve with loss of renal function would be of great relevance in clinical practice.
IgAN is an important cause of kidney failure. It should be suspected in patients with a macroscopic hematuria, especially if there is a febrile illness, or with asymptomatic hematuria and/or proteinuria. The disease is characterized by glomerular immunodeposits enriched for Gd-IgA1 glycoforms and for IgG autoantibodies with specificity for the IgA1 with galactose-deficient O-glycans. In few words, the basis of the disease result from the formation of circulating immune complexes comprised of Gd-IgA1 bound primarily to IgG autoantibody. These immune complexes accumulate in the mesangium, leading to inflammation and progressive scarring with loss of kidney function.
Currently, there is no specific treatment for the disease and it is hoped that with advances in understanding its pathogenesis we will be able to have an earlier diagnosis, more appropriate treatment and better survival for patients.
Declaration of conflict of interest:
The authors declare no conflicts of interest between the investigators and the patients, and other institutions.
Funding agency name:
None.
Indication of authors' contribution:
Maria Virginia Pereira Gomide Freitas and Maria Goretti Moreira Guimarães Penido were responsible for the research idea, study design, data acquisition, supervision or mentorship and article writing.














Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org