Clinical Case Reports and Clinical Study
OPEN ACCESS | Volume 13 - Issue 1 - 2026
ISSN No: 2766-8614 | Journal DOI: 10.61148/2766-8614/JCCRCS


Naman Sharma
Internal Medicine, Rochester General Hospital · Rochester, USA.
*Corresponding author: Naman Sharma, Internal Medicine, Rochester General Hospital · Rochester, USA.
Received Date: December 20, 2021
Accepted Date: January 03, 2022
Published Date: January 12, 2022
Citation: Naman Sharma, (2022) “Donor cell Leukemia after Cord Blood Transplantation for IPEX Syndrome.” Clinical Case Reports and Clinical Study, 6(1); DOI: http;//doi.org/12.2022/1.123.
Copyright: © 2022 Naman Sharma. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
,
Case
A 28 year male was diagnosed with the rare IPEX syndrome at age 17 after a prolonged workup which ultimately identified a hallmark FOXP3 (Fork-head box P) mutation. IPEX syndrome is potentially life- threatening due to hemizygous mutations in FOXP3 that prevent immunological tolerance by thymus- derived regulatory T cells (Tregs). It presents classically in infant males with a triad of enteropathy, dermatitis and autoimmune endocrionopathy, but milder forms may present later. While immunosuppressive therapy is used, IPEX is currently only curative by allogeneic HSCT1. Without a suitably matched sibling or unrelated donor, the patient underwent double cord blood transplant (CBT) at age 20, after pre-conditioning with treosulfan, fludarabin, and 2 Gy total body irradiation. Post transplant chimerism revealed 95% contribution from a single cord from a female donor. He developed mild GVHD managed with sirolimus and steroids. The last chimerism available almost 8 years post- transplant showed 100 % contribution from the female donor in CD3, CD33, and CD 56 fractions. There was no signal detected from the patient or the other cord graft.
Later that year, he developed worsening thrombocytopenia. Bone marrow biopsy showed tri- lineage dysplasia with chromosome 6q and 7p deletions that were donor derived based on 499/500 cells showing X,X chromosomes by FISH. He began 5-azacitidine therapy and became transfusion dependent for red cells and platelets. A peripheral blood smear 6 months later revealed 8% blasts and dysgranulopoiesis (Figure 1A). Bone marrow aspirate showed WHO 3+ marrow fibrosis. Chromosome G- banding analysis again confirmed donor-derived female karyotype in all 20 metaphases, along with derivative chromosomal translocation between the short arm of chromosome 6 (6p) and long arm of chromosome 7 (7q). This clonal abnormality resulted in 6q and 7p deletions (Figure 1B-D). Additional mutations detected by next generation sequencing using the Ilumina TruseqTM myeloid panelTM included one in B-Raf (c.1803A>T, p.Lys601Asn, variant allele frequency (VAF) 41% and ASXL1(c.2464_2467delACAT, p.Thr822Ter, VAF 45%).
A second allogeneic HCT was recommended. Once again, no suitably matched unrelated donors were identified in the National Marrow Donor Program registry. His female relatives were FOXP3 mutation carriers, and he had no other related donors available. A suitable cord was identified that contained 5.33 X 107 nucleated cells/Kg and 4.89 X 105 CD 34+ cells/Kg. His haploidentical sister, a FOXP3 mutation carrier, was worked-up as a backup donor.
Eight months after the diagnosis of MDS, while being worked up for a second CBT, he presented with confusion, lethargy, abdominal pain, hypoxia and hypotension. CT showed diffuse anasarca and diffuse edema of the mesenteric and pelvic fat, bilateral pleural effusions, enlarged mediastinal and axillary lymph nodes and ground glass opacities in the lungs. White blood cell count was 85,900/μL (24% blasts), hematocrit 29%, and platelets 20,000/μL. This progression to acute myelogenous leukemia was treated with daunorubicin and cytarabine. He was given broad spectrum anti-fungal and anti-bacterial agents, but multifocal pneumonia and sepsis progressed. He succumbed to disseminated fungal infection.
Discussion
IPEX syndrome is a rare auto-immune disorder due to a mutation in the FOXP3 gene responsible for differentiation and regulation of Tregs. It presents classically in infant males with a triad of enteropathy, dermatitis and autoimmune endorinopathy and is potentially fatal without treatment with immunosuppressants. HSCT is the only potential cure1.
Conventionally bone marrow and peripheral blood have been used as the stem cell source for allografting, but in those who have no suitable matched related or unrelated donor and no haploidentical donor options, umbilical cord blood transplantation (UCBT) can be used with potential benefits of lower incidence of post-transplant complications like acute and chronic GVHD 2 without increase in the relapse rates. In this case, where transplant was performed a decade ago, haploidentical transplants were performed less commonly; hence the use of cord blood donors. IPEX syndrome in itself is not associated with secondary malignancies.
When transplant is performed for hematologic malignancies, relapse usually occurs in host stem cells with phenotypic and molecular features that mirror the original malignancy. This case highlights the rare, de-novo leukemic transformation within the donor cell compartment, referred to as Donor Cell Leukemia. First described in
19713 , the true incidence is unknown due to complexity in making the diagnosis, non- standardized methods for confirming the diagnosis, and heterogeneity in reported cases. The incidence of DCL is thought to be around 0.1- 5% 4,5 and the first case after UBCT was reported in 20056 . Since then DCL has been rarely reported, and in total only around 10 cases arising in UCBT graft. To our knowledge, this is the first reported case of DCL after UCBT for IPEX, a non- malignant disorder.
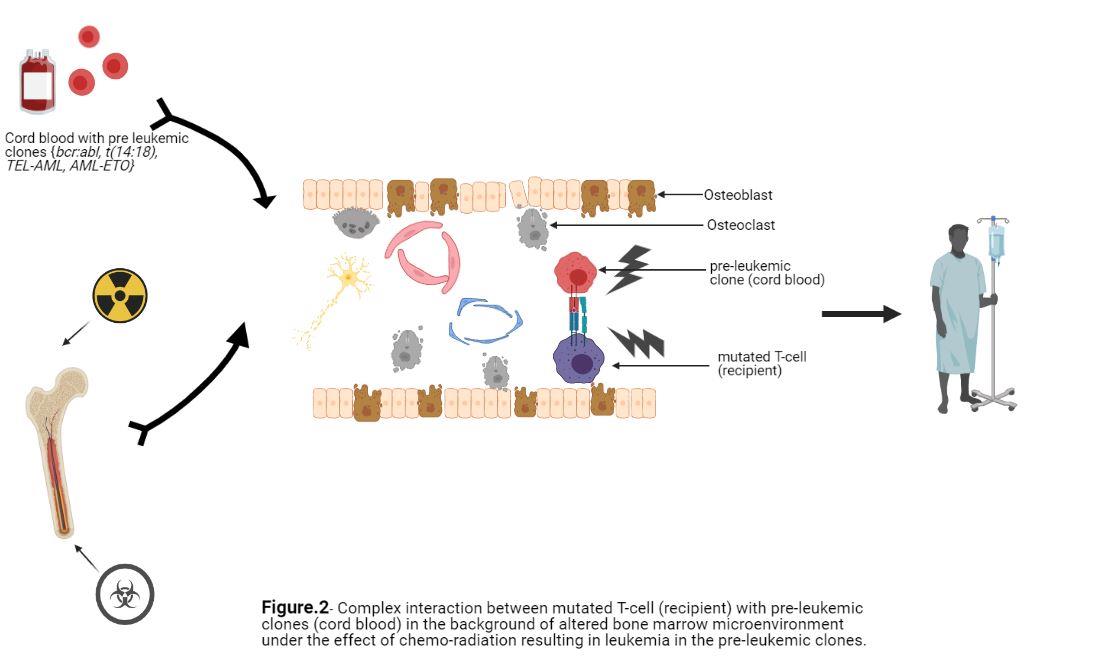
The pathogenesis of DCL is not clearly understood and is thought to be the result of interplay between various mechanisms; mutation in transcription factors (e.g. Enhancer Binding Protein family/ EBP) and transduction proteins (e.g.- Janus Kinase family) 7 resulting in aberrant cell-cell signaling in a marrow environment affected by prior chemotherapy and radiation exposures, sustained antigenic stimulation due to host- donor minor histocompatibility differences, fusion of grafted cells with residual malignant cells 8 , and errors in DNA repair due to replicative stress 11. Presence of perleukemic clones in normal donors (bcr-abl mRNA in ~30 % 10, t (14:18) detectable in 24 % 11 and PCR screening of cord blood samples revealing ~1 % TEL-AML1 and 0.2 % AML-ETO mutation12 has been noted. However, progression to fulminant disease in the donor is very rare, and in most cases, there is no documentation of a preleukemic clone. In our case, it is possible any residual mutated and activated T cell from the recipient after the CBT could effect the marrow milieu for donor cord blood stem cells via cytokine production and other means. None of these proposed mechanisms have been extensively studied or documented, but they point out possible complex interactions of pre-leukemic clones with altered bone marrow microenvironment, followed by additional hits resulting in fulminant disease, supporting the “multiple hit” hypothesis (Figure 2)13. Overall, all these mechanisms highlighted are not mutually exclusive and may occur simultaneously.
Making a diagnosis of DCL can be difficult. The first suspicion of DCL arises when an emerging blast percentage is associated with complete or predominantly donor chimerism in unfractionated leukocytes or the relevant (usually myeloid, lymphoid) microfluorimetry-sorted leukocyte fraction.
There are various techniques available to demonstrate donor cell transformation with certain limitations and pitfalls. For example: XY-FISH and chromosomal analysis is only applicable to sex-mismatched transplant and because leukemic cells can be genetically unstable and Y chromosome can be lost, this should not be used as the sole criterion14. Using VNTR/ STR (variable number tandem repeat/short tandem repeat) analysis, identification of a unique donor allele can serve as an informative marker, and presence of the non-shared allele in the leukemic blast can be considered as a donor origin of leukemia, but stochastic amplification (especially with VNTR) can cause allele drop-out and therefore decrease the detection threshold 15. In face of nuances in the test interoperation and without standardized testing and guidelines, a combination of tests is preferred. In the case described here, all cells on karyotype were X;X and these cells also carried the translocation between chromosomes 6 and 7.
Our patient developed MDS 8 years after UBCT that progressed to donor- derived AML which falls within previous reported timeframe for DCL at a median of 31 months (range 2-312 months, longer if preceded by antecedent MDS 13 ). One another interesting molecular finding in our patient is the presence of a B-raf mutation (B-raf c.1803A>T (p.Lys601Asn) with allele frequency of 45%, not previously found in polymorphism databases or associated with myeloid malignancies. Most mutations at this hotspot are Lys601Glu, and B-raf mutations are rarely associated with myeloid leukemias or with the IPEX syndrome. As noted, a CHIP- related ASXL1 mutation was also present which has been recognized as a novel risk factor for DCL 16.
Similar to therapy-related AML, the prognosis of DCL is extremely poor with median survival of 5.5 months 17. Reinduction therapies are administered as a bridge to second HSCT from an alternative donor if possible.
Overall, DCL is a very intriguing phenomenon that has been increasingly identified and diagnosed and can serve as a model for the prospective study of leukemogenesis. The entity should be kept in mind when MDS/leukemia develops after HSCT and phenotypic features differ from the original disease, if the relapse occurs late, or if the transplant was for a non-malignant disease that has no known association with leukemia as was the case here. With molecular techniques for chimerism, DCL is being increasingly diagnosed. Despite all the advancements made, there remains a dearth of evidence due to lack of large scale studies and guidelines for clinical management.
Along with diagnostic and treatment challenges, DCL also presents ethical concerns 17 regarding additional responsibilities of the physicians and the donor registries towards the donor. Unlike adult donors, there is no specific policy in the case of UBCT, with no opportunity for donor tracing. Notifying childhood or adult donors in case of adverse events in the recipients may result in feelings of guilt along with additional anxiety regarding their own fate without clear benefit and unknown impact on their social, financial and psychological life.
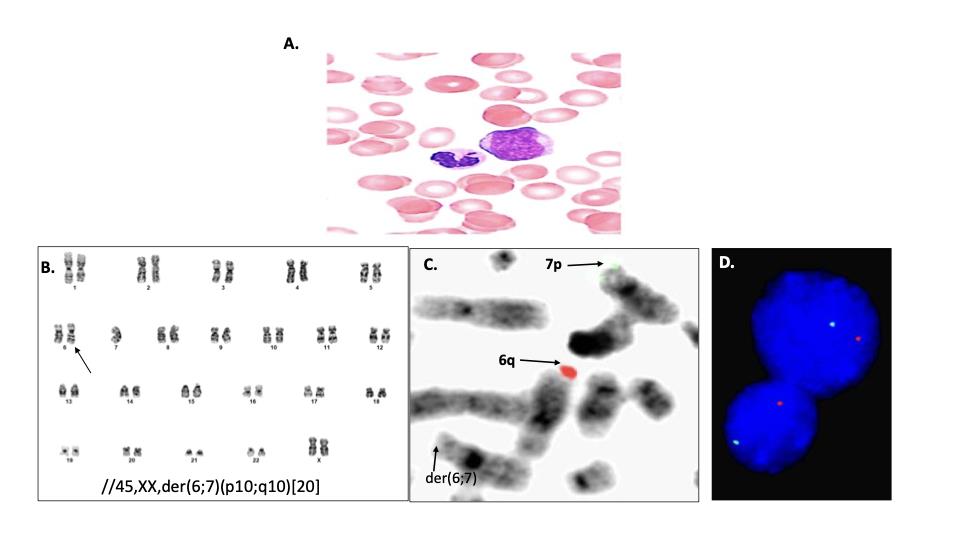
Figure 1- Peripheral blood and some marrow cytogenetic analysis. (A) blood film showing myeloblast with agranular neutrophil (x1000, Wright-Giemsa stain). (B) G- banding analysis identified a female karyotype containing a derivative chromosome (arrow) resulting from a whole arm translocation between the short arm of chromosome 6 and long arm of chromosome 7. (C) Metaphase FISH showed absence of 6q and 7p signals (6;7). (D) Interphase FISH confirmed 6q and 7p deletions in 77% of cells analyzed (nuc ish(subtel6qx1,subtel7px1)[154/200]).















Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org