Aditum Journal of Clinical and Biomedical Research
OPEN ACCESS | Volume 8 - Issue 1 - 2026
ISSN No: 2993-9968 | Journal DOI: 10.61148/2993-9968/AJCBR


L. Sarvananda1*, Janitha H. Meepearachchi2, Amal D Premarathne3
1Molecular Nutritional and Biochemistry Laboratory, University of Peradeniya, Peradeniya, Sri Lanka.
2Department of Biotechnology, Faculty of Science, Horizon Campus, Malabe, Sri Lanka.
3School of Natural Sciences and Health, Tallinn University, Narvamnt 29, 10120 Tallinn, Estonia.
*Corresponding Author: L. Sarvananda, Molecular Nutritional and Biochemistry Laboratory, University of Peradeniya, Peradeniya, Sri Lanka.
Received: April 17, 2021
Accepted: April 20, 2021
Published: April 26, 2021
Citation: L. Sarvananda, Janitha H. Meepearachchi and Amal D Premarathne. (2021) “A Brief of Nucleic Acid Metabolism”, Aditum Journal of Clinical and Biomedical Research, 3(1); DOI: http;//doi.org/04.2021/1.1024.
Copyright: © 2021 L. Sarvananda. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Biosynthesis of Purine Nucleotides and their inter conversion –regulation of biosynthesis:
Origin of the ring atoms of purines obtained from isotopic experiments with 14C- or 15N labeled precursors. Formate is supplied in the form of N10- formyltetrahydrofolate. N1 of Purines arises from the amino group of aspartate C2 and C8 originate from formate N3 and N9 are contributed by the amide group of glutamine C4,C5, and N7 are derived from glycine (strongly suggesting that this molecule is wholly incorporated into the purine ring) and C6 comes from HCO3-CO2.
Introduction:
Biosynthesis of Purine Nucleotides and their inter conversion –regulation of biosynthesis:
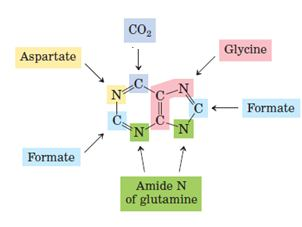
Origin of the ring atoms of purines obtained from isotopic experiments with 14C- or 15N labeled precursors. Formate is supplied in the form of N10- formyltetrahydrofolate. N1 of Purines arises from the amino group of aspartate C2 and C8 originate from formate N3 and N9 are contributed by the amide group of glutamine C4,C5, and N7 are derived from glycine (strongly suggesting that this molecule is wholly incorporated into the purine ring) and C6 comes from HCO3-CO2.
Steps: Ribose 5-phosphate produced in the hexose monophosphate shunt of carbohydrate metabolism is the starting material for purine nucleotide synthesis. It reacts with ATP to form phosphoribosyl pyrophosphate (PRPP). Glutamine transfers its amide nitrogen to PRPP to replace pyrophosphate and produce 5-phosphoribosylamine. The enzymes PRPP glutamyl amidotransferases is controlled by feedback inhibition of nucleotides (IMP, AMP, and GMP). This reaction is the committed step in purine nucleotide biosynthesis.
Phosphoribosylaminere acts with glycine in the presence of ATP to form glycinamide ribosyl 5-phosphate or glycinamide ribotide (GAR). N10-Formyl tetrahydrofolate donates the formyl group and the product formed is formylglycinamide ribosyl 5-phosphate. Glutamine transfers the second amido amino group to produce formylglycinamidine ribosyl 5-phosphate.
The imidazole ring of the purine is closed in an ATP dependent reaction to yield 5-aminoimidazole ribosyl S-phosphate. Incorporation of CO2 (carboxylation) occurs to yield aminoimidazole carboxylate ribosyl 5-phosphate. This reaction does not require the vitamin biotin and/or ATP which is the case with most of the carboxvlation reactions. Aspartate condenses with the product in reaction 7 to form aminoimidazole 4- succinyl carboxamide ribosyl 5-phosphate. Adenosuccinate lyase cleaves off fumarate and only the amino group of aspartates is retained to yield aminoimidazole4 –carboxamide ribosyl 5- phosphate. N1O-Formyl tetrahydrofolate donates a one-carbon moiety to produce formaminoimidazole 4-carboxamide ribosyl 5-phosphate. With this reaction, all the carbon and nitrogen atoms of purine ring are contributed by the respective sources. The final reaction catalysed by cyclohydrolase leads to ring closure with an elimination of water molecule. The product obtained is inosine monophosphate (IMP), the parent purine nucleotide from which other purine nucleotides can be synthesized.
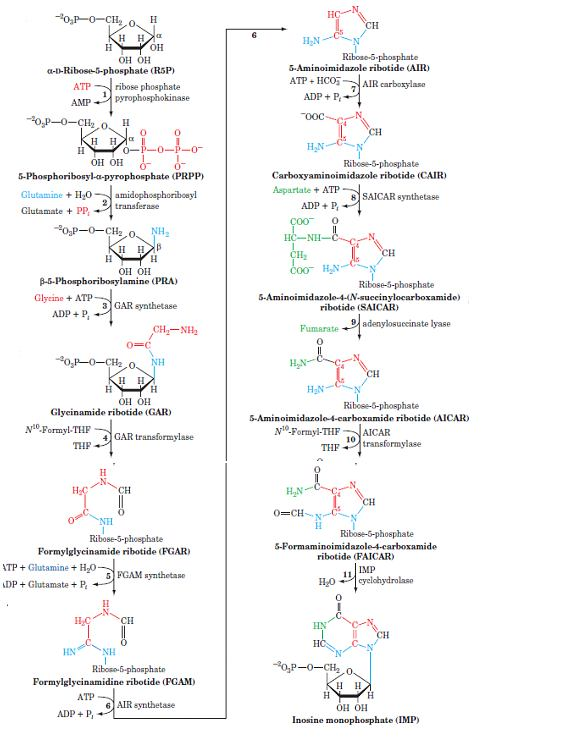
Regulation: The purine nucleotide synthesis is well coordinated to meet the cellular demands. The intracellular concentration of PRPP regulates purine synthesis to a large extent. This, in turn, is dependent on the availability of ribose 5-phosphate and the enzyme PRPP synthetase. PRPP glutamyl amidotransferase is controlled by a feedback mechanism by purine nucleotides. That is, if AMP and CMP are available in adequate amounts to meet the cellular requirements their synthesisis turned off at the amidotransferase reaction.
Another important stage of regulation is in the conversion of IMP to AMP and CMP. AMP inhibits adenylsuccinate synthetase while CMP inhibits IMP dehydrogenase. Thus, AMP and CMP control their respectives ynthesis from IMP by a feedback mechanism.
Biosynthesis of pyrimidine nucleotides and their inter conversion - regulation of biosynthesis:
Nucleotides play a variety of important roles in all cells. They are the precursors of DNA and RNA. They are essential carriers of chemical energy a role primarily of ATP and to some extent GTP. They are components of the cofactors NAD, FAD, S-adenosylmethionine, and coenzyme A, as well as of activated biosynthetic intermediates such as UDP-glucose and CDP diacylglycerol.
Two types of pathways lead to nucleotides in that, one is De novo synthesis of nucleotides begins with their metabolic precursors: Amino acids, ribose 5-phosphate, CO2, and NH3 in Liver. Another pathway is Salvage pathways recycle the free bases and nucleosides released from nucleic acid breakdown in RBC, Brain.
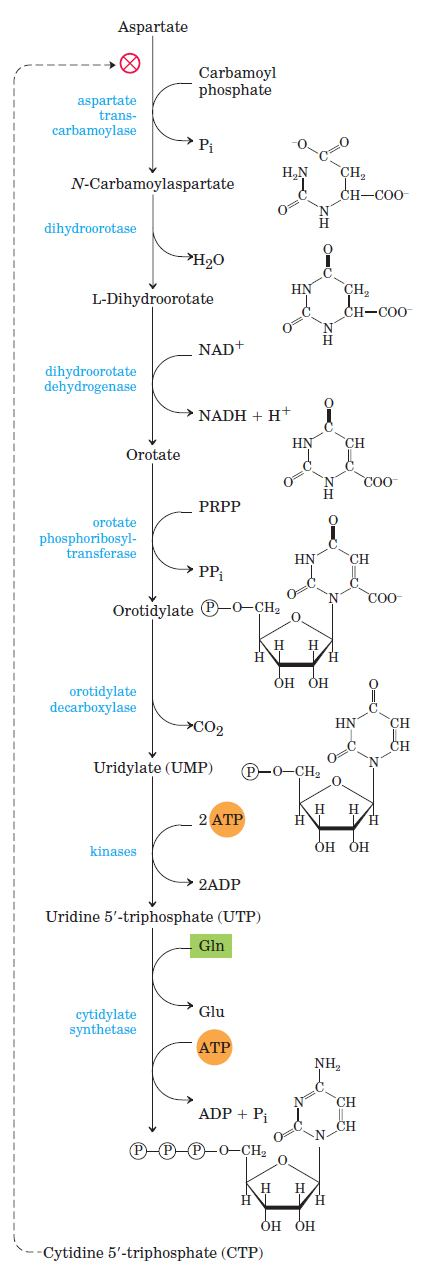
Pyrimidine Nucleotides Are Made from Aspartate, PRPP, and Carbamoyl Phosphate. The common pyrimidine ribonucleotides are cytidine 5 monophosphate (CMP; cytidylate) and uridine 5 monophosphate (UMP; uridylate), which contain the pyrimidines cytosine and uracil. De novo pyrimidine nucleotide biosynthesis proceeds in a somewhat different manner from purine nucleotide synthesis; the six-membered pyrimidine ring is made first and then attached to ribose 5- phosphate.
Required in this process is carbamoyl phosphate, also an intermediate invthe urea cycle Carbamoyl phosphate reacts with aspartate to yield N-carbamoylaspartate in the first committed step of pyrimidine biosynthesis. This reaction is catalyzed by aspartate transcarbamoylase De novo synthesis of pyrimidine nucleotides: biosynthesis of UTP and CTP via orotidylate. The pyrimidine is constructed from carbamoyl phosphate and aspartate. The ribose 5-phosphate is then added to the completed pyrimidine ring by orotate phosphoribosyl transferase.
Regulation: Regulation of the rate of pyrimidine nucleotide synthesis in bacteria occurs in large part through aspartate transcarbamoylase (ATCase), which catalyzes the first reaction in the sequence and is inhibited by CTP, the end product of the sequence. The bacterial ATCase molecule consists of six catalytic subunits and six regulatory subunits. The catalytic subunits bind the substrate molecules, and the allosteric subunits bind the allosteric inhibitor, CTP. The entire ATCase molecule, as well as its subunits, exists in two conformations, active and inactive. When CTP is not bound to the regulatory subunits, the enzyme is maximally active. As CTP accumulates and binds to the regulatory subunits, they undergo a change in conformation. This change is transmitted to the catalytic subunits, which then also shift to an inactive conformation. ATP prevents the changes induced by CTP.
Importance of Thymidylate synthase:
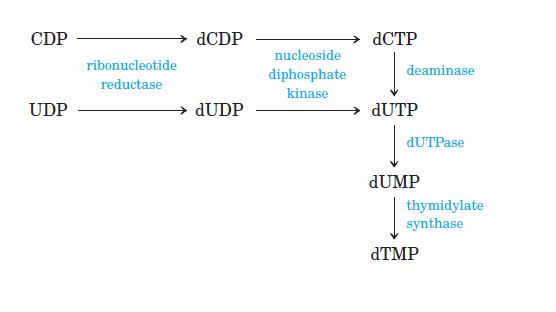
Thymidylate Synthase dTMP is synthesized from dUMP by thymidylate synthase (TS) with N5, N10-methylenetetrahydrofolate (N5, N10-methylene- THF) as the methyl donor:
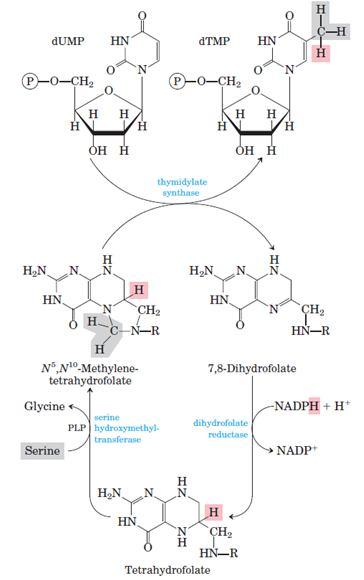
Conversion of dUMP to dTMP by thymidylate synthase and dihydrofolate reductase.
Serine hydroxymethyltransferase is required for regeneration of the N5, N10- methylene form of tetrahydrofolate. In the synthesis of dTMP, all three hydrogens of the added methyl group are derived from N5, N10 methylene tetrahydrofolate (pink and gray).
Other Pathways of Purine Nucleotide Formation:
Salvage pathways:
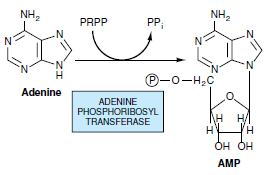
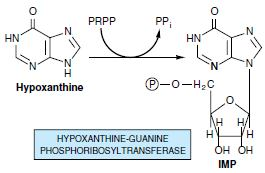
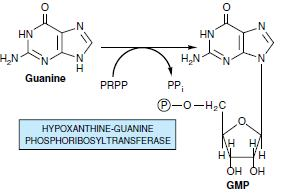
Most cells have an active turnover of many of their nucleic acids (particularly some types of RNA) which, through degradative processes, result in the release of adenine, guanine, and hypoxanthine. These free purines are reconverted to their corresponding nucleotides through salvage pathways. In contrast to the de novo purine nucleotide synthesis pathway, which is virtually identical in all cells, salvage pathways are diverse in character and distribution. In mammals, purines are, for the most part, salvaged by two different enzymes. Adenine phosphoribosyltransferase (APRT) mediates AMP formation through the transfer of adenine to PRPP with the release of PPi:
Biosynthesis of Deoxyribonucleotides Nucleotides:
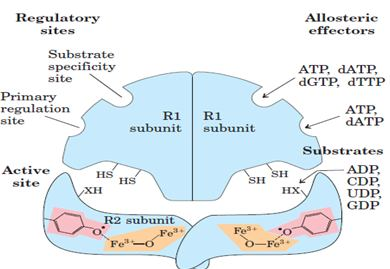
Deoxyribonucleotides are synthesized from their corresponding ribonucleotide by reduction of their C2’ Position rather than their denovo synthesis from deoxyribose containing precursors. The reaction is catalyzed by ribonucleotide reductase, and the substrate is ribonucleotide diphosphates. There are 3 classes of ribonucleotide reductases:
Class I: They have a tyrosyl radical that is stabilized by an oxobridged binuclear Fe III complex. They are widely distributed among prokaryotes.
Class II: They utilize a deoxy adenosyl cobalamine as a cofactor (coenzyme B- 12). They are widely distributed among prokaryotes
Class III: They contain [3Fe-4S] cluster and require SAM and NADPH for activity.they occurs in prokaryotes that grow anaerobically.
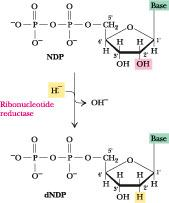
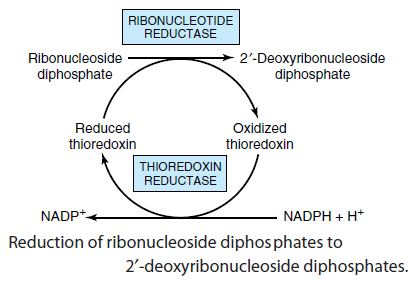
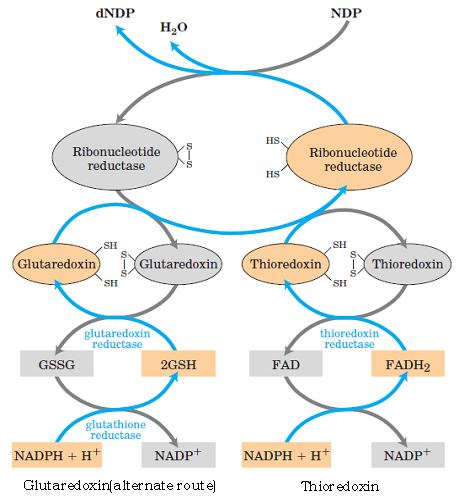
The reduction of the D-ribose portion of a ribonucleoside diphosphate to 2’- deoxy-D-ribose requires a pair of hydrogen atoms, which are ultimately donated by NADPH via an intermediate hydrogen-carrying protein, thioredoxin. Groups that carry hydrogen atoms from NADPH to the ribonucleoside diphosphate. Its oxidized (disulfide) form is reduced by NADPH in a reaction catalyzed by thioredoxin reductase, and reduced thioredoxin is then used by ribonucleotide reductase to reduce the nucleoside diphosphates (NDPs) to deoxyribonucleoside diphosphates (dNDPs). A second source of reducing equivalents for ribonucleotide reductase is glutathione (GSH). Glutathione serves as the reductant for a protein closely related to thioredoxin glutaredoxin, which then transfers the reducing power to ribonucleotide reductase.
Biosynthesis of thymidylate:
Biosynthesis of Coenzymes Nucleotides:
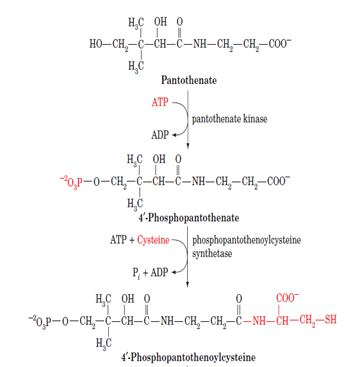
Biosynthesis of coenzyme A:
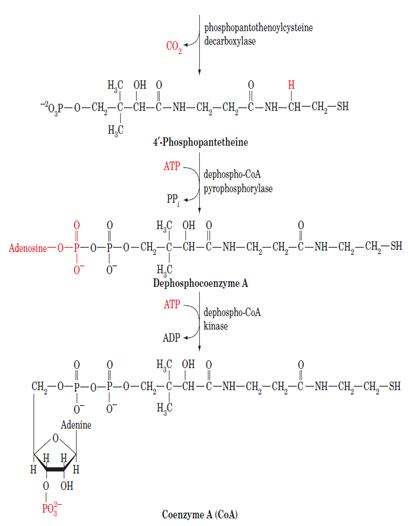
Coenzyme A is synthesized in mammalian cells according to the pathway diagrammed below. In Pantothenate, an essential vitamin, is phosphorylated by pantothenate kinase and then coupled to cysteine, the future business end of CoA, by phosphopantothenoylcysteine synthetase. After decarboxylation by phosphopantothenoylcysteine decarboxylase, the resulting 4-phosphopantethiene is coupled to AMP in a pyrophosphate linkage by dephospho- CoA pyrophosphorylase and then phosphorylated at its adenosine 3¿ OH group by dephospho-CoA kinase to form CoA. The latter two enzymatic activities occur on a single protein.
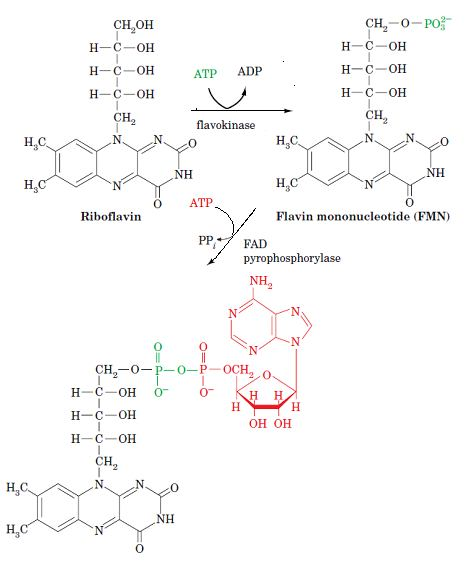
Biosynthesis of FAD:
FAD is synthesized from riboflavin in a two-reaction pathway. First, the 5’OH group of riboflavin’s ribityl side chain is phosphorylated by flavokinase, yielding flavin mononucleotide (FMN; not a true nucleotide since its ribityl residue is not a true sugar).
FAD may then be formed by the coupling of FMN and ATP-derived AMP in a pyrophosphate linkage in a reaction catalyzed by FAD pyrophosphorylase. Both of these enzymes are widely distributed in nature.
Chemical Inhibition of The Biosynthesis of Nucleic Acid Precursors:
Methotrexate act as an anticancer agent: The growth of cancer cells is not controlled in the same way as cell growth in most normal tissues. Cancer cells have greater requirements for nucleotides as precursors of DNA and RNA, and consequently are generally more sensitive than normal cells to inhibitors of nucleotide biosynthesis. A growing array of important chemotherapeutic agents for cancer and other diseases act by inhibiting one or more enzymes in the nucleotide biosynthesis pathways.
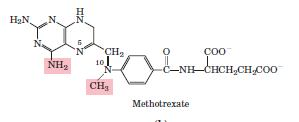
Methotrexate is an inhibitor of dihydrofolate reductase. This folate analog acts as a competitive inhibitor; the enzyme binds methotrexate with about 100 times higher affinity than dihydrofolate.
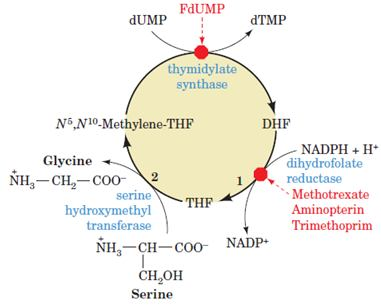
Antifolates: Are Anticancer Agents Inhibition of DHFR quickly results in all of a cell’s limited supply of THF being converted to DHF by the thymidylate synthase reaction. Inhibition of DHFR therefore not only prevents dTMP synthesis, but also blocks all other THF-dependent biological reactions such as the synthesis of purines, methionine, and, indirectly, histidine. DHFR therefore offers an attractive target for chemotherapy.
Degradation of Purine:
Degradation of Purines and Produces Uric Acid Purine nucleotides are degraded by a pathway in which they lose their phosphate through the action of 5’- nucleotidase. Adenylate yields adenosine, which is deaminated to inosine by adenosine deaminase, and inosine is hydrolyzed to hypoxanthine (its purine base) and D-ribose. Hypoxanthine is oxidized successively to xanthine and then uric acid by xanthine oxidase, a flavoenzyme with an atom of molybdenum and four ironsulfur centers in its prosthetic group. Molecular oxygen is the electron acceptor in this complex reaction.
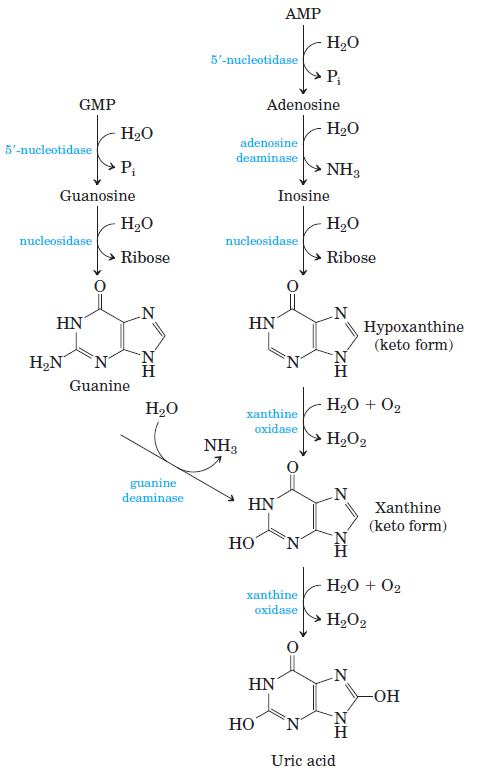
GMP catabolism also yields uric acid as end product. GMP is first hydrolyzed to guanosine, which is then cleaved to free guanine. Guanine undergoes hydrolytic removal of its amino group to yield xanthine, which is converted to uric acid by xanthine oxidase.
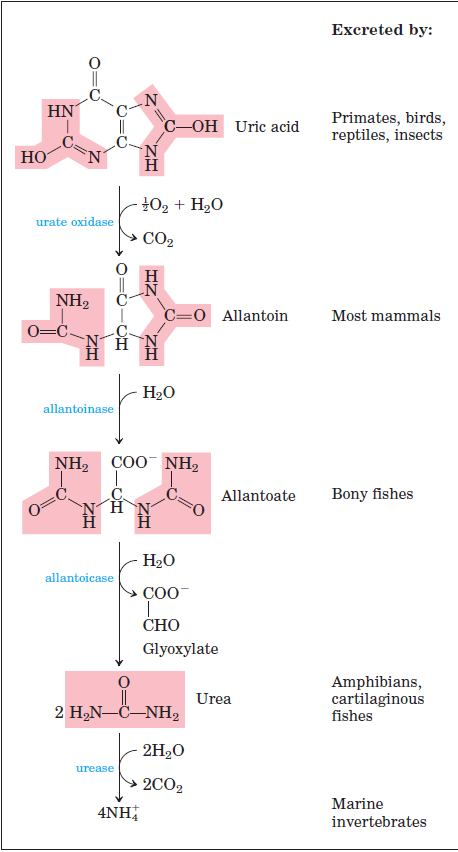
Uric acid is the excreted end product of purine catabolism in primates, birds, and some other animals. A healthy adult human excretes uric acid at a rate of about 0.6 g/24 h; the excreted product arises in part from ingested purines and in part from turnover of the purine nucleotides of nucleic acids. In most mammals and many other vertebrates, uric acid is further degraded to allantoin by the action of urate oxidase.
Purine nucleotide cycle:
The deamination of AMP to IMP, when combined with the synthesis of AMPfrom IMP has the effect of deaminating aspartate to yield fumarate. John Lowenstein demonstrated that this purine nucleotide cycle has an important metabolic role in skeletal muscle. An increase in muscle activity requires an increase in the activity of the citric acid cycle. This process usually occurs through the generation of additional citric acid cycle intermediates.
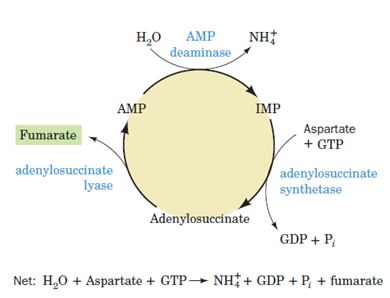
Muscles, however, lack most of the enzymes that catalyze these anaplerotic (filling up) reactions in other tissues. Rather, muscle replenishes its citric acid cycle intermediates as fumarate generated in the purine nucleotide cycle. The importance of the purine nucleotide cycle in muscle metabolism is indicated by the observation that the activities of the three enzymes involved are all severalfold higher in muscle than in other tissues. In fact, individuals with an inherited deficiency in muscle AMP deaminase (myoadenylate deaminase deficiency) are easily fatigued and usually suffer from cramps after exercise.
Degradation of Pyrimidines:
Animal cells degrade pyrimidine nucleotides to their component bases. These reactions, like those of purine nucleotides, occur through dephosphorylation, deamination, and glycosidic bond cleavages. The resulting uracil and thymine are then broken down in the liver through reduction rather than by oxidation, as occurs in purine catabolism.The end products of pyrimidine catabolism, β -alanine and β-aminoisobutyrate, are amino acids and are metabolized as such. They are converted, through transamination and activation reactions, to malonyl- CoA and methylmalonyl-CoA for further utilization.
Disorders Associated with Their Metabolism – Gout:
Gout is a disease of the joints caused by an elevated concentration of uric acid in the blood and tissues. The joints become inflamed, painful, and arthritic, owing to the abnormal deposition of sodium urate crystals. The kidneys are also affected, as excess uric acid is deposited in the kidney tubules. Gout occurs predominantly in males. Its precise cause is not known, but it often involves an underexcretion of urate. A genetic deficiency of one or another enzyme of purine metabolism may also be a factor in some cases.
Treatment: Gout is effectively treated by a combination of nutritional and drug therapies. Foods especially rich in nucleotides and nucleic acids, such as liver or glandular products, are withheld from the diet. Major alleviation of the symptoms is provided by the drug allopurinol, which inhibits xanthine oxidase, the enzyme that catalyzes the conversion of purines to uric acid.
Allopurinol is a substrate of xanthine oxidase, which converts allopurinol to oxypurinol (alloxanthine). Oxypurinol inactivates the reduced form of the enzyme by remaining tightly bound in its active site. When xanthine oxidase is inhibited, the excreted products of purine metabolism are xanthine and hypoxanthine, which are more watersoluble than uric acid and less likely to form crystalline deposits.
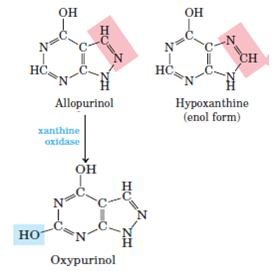
Allopurinol, an inhibitor of xanthine oxidase. Hypoxanthine is the normal substrate of xanthine oxidase. Only a slight alteration in the structure of hypoxanthine (shaded pink) yields the medically effective enzyme inhibitor allopurinol. At the active site, allopurinol is converted to oxypurinol, a strong competitive inhibitor that remains tightly bound to the reduced form of the enzyme
Lesch-Nyhan Syndrome:
Only males are affected by this It is X linked recessive defect. Enzyme deficiency: Hypoxanthine guanine phosphoribosyltransferase. The enzyme is almost absent and leads to increased purines salvage pathway from PRPP. Symptoms are Severe gout, renal failure, poor growth, spasticity and tendency for self-mutilation
Oroticaciduria:
Excretion of orotic acid in urine is known as orotic aciduria. The orotic aciduria that accompanies Reye’s syndrome probably is a consequence of the inability of severely damaged mitochondria to utilize carbamoyl phosphate, which then becomes available for cytosolic over production of orotic acid. Type I orotic aciduria reflects a deficiency of both orotate phosphoribosyltransferase and orotidylate decarboxylase; the rarer type II orotic aciduria is due to a deficiency only of orotidylate decarboxylase.
Xanthinuria:
Hypouricemia and increased excretion of hypoxanthine and xanthine are associated with xanthine oxidase deficiency due to a genetic defect or to severe liver damage. Patients with a severe enzyme deficiency may exhibit xanthinuria and xanthine lithiasis.
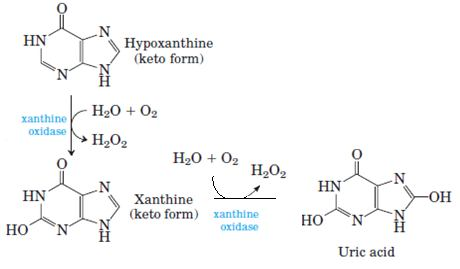
Inheritence: Autosomal recessive. Enzyme deficiency: Xanthine oxidase. Xanthine oxidases blocks the oxidation of hypoxanthine and xanthine to uric acid. Causes are Hypouricemia and xanthine lithiasis.
SCID:
If, however, several molecules of the biologically inert polymer polyethylene glycol (PEG) are covalently linked to surface groups on ADA, the resulting PEG–ADA remains in the blood for 1 to 2 weeks, thereby largely resuscitating the SCID victim’s immune system.





Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org