Aditum Journal of Clinical and Biomedical Research
OPEN ACCESS | Volume 8 - Issue 1 - 2026
ISSN No: 2993-9968 | Journal DOI: 10.61148/2993-9968/AJCBR


Kulvinder Kochar Kaur 1*, Gautam Allahbadia 2, Mandeep Singh 3
1Kulvinder Kochar Kaur, Scientific Director, Dr Kulvinder Kaur Centre for Human Reproduction 721, GTB. Nagar, Jalandhar-144001, Punjab, India
2Ex-Rotunda-A Centre for Human Reproduction 672, Kalpak Garden,Perry Cross Road, Near Otter’s Club,Bandra(W).
3Consultant Neurologist Swami Satyanand Hospital Near Nawi Kachehri,Baradri, Ladowali road,Jalandhar Punjab.
*Corresponding Author: Kulvinder Kochar Kaur, Kulvinder Kochar Kaur, Scientific Director, Dr Kulvinder Kaur Centre for Human Reproduction 721, GTB. Nagar, Jalandhar-144001, Punjab, India.
Received: March 03, 2021
Accepted: April 08, 2021
Published: April 14, 2021
Citation: Kochar Kaur.K, Allahbadia.G, Singh.M. (2021) “Would it be advantageous utilizing beta cell therapies over immunotherapies for avoidance of Type 1 Diabetes-A Systematic Review on the role of beta cells in etiopathogenesis of Type 1 Diabetes along with treatments targeting beta cells or combination therapy would be better”, Aditum Journal of Clinical and Biomedical Research, 1(2); DOI:http;//doi.org/04.2021/1.1010.
Copyright: © 2021 Kulvinder Kochar Kaur. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Earlier we had reviewed various aspects of Type 1 Diabetes(T1D)(,its etiopathogenesis,various immunotherapies used and how we could try to obviate the need of insulin ,role of empagliflozin addition ,role of extracellular vesicles(ECV’s) in treating complications associated with T1D ,role of gut microbiota and early life feeding,genes responsible (unpublished ),epigenetics in Diabetic Kidney Disease(DKD),The etiopathogenesis of T1D despite the earlier belief that it represents an autoimmune diseases with continuing autoimmune modulated damage of pancreatic β cells. Thus Here we conducted a systematic review utilizing search engine pubmed,google scholar ;web of science ;embase; Cochrane review library utilizing the MeSH terms like; Type 1 Diabetes(T1D ;beta cell in etiopathogenesis of T1D;Immunotherapies ;role of Unfolded proteins response(UPR);role of senescent β cells ; Role of Type 1 Interferon ;DNA methylation;PDL1 ;Little insulin generation by αcells besides glucagon ;other endocriner cells of pancreas ;Role of autophagy;other mechanisms like apoptosis ;necrosis in β cell demise ; endoplasmic reticulum (ER)stress ;Terminal UPR;Advanced UPR;EM alterations in mitochondria of islet β cell;Endotype;heterogeneity in T1D; of latent autoimmune Diabetes in Adults(LADA) ;Immunotherapies ; β cell therapies ;combination of 2 therapies;DDR;Senolytics;Bcl2;Bcl –XL; circulating cell free DNA (cfDNA) ; Histone mimic suppression of inflammation;BET Inhibitors(Molibresib); Epigenetics modulation of macrophages and β cells; Tauroursodeoxycholic acid(TUDCA), Verapamil(TXNIP inhibitor) Imatinib (IRE1α- ABL inhibitor from 1950 to 2021 till date.We found a total of 300 articles out of which we selected 135 articles for this review.No meta-analysis was done.Thus we have discussed the different pathways that influence the β cell impairments .Various etiologies like UPR ,SASP are reviewed along with pathways for β cell targeted therapieslike Verapamil([thioredoxin –interacting protein(TXNIP)] inhibitor) Imatinib (IRE1α[inositol requiring enzyme -1 alpha(IRE1α)],- Abelson tyrosine protein kinase (ABL ), BET Inhibitors(Molibresib); Tauroursodeoxycholic acid(TUDCA).Further the existing queries that still need to be resolved are duiscussed .This was we might be able to shorten the gap in T1D etiology as well as maximize the potential of these therapies or existing immunotherapies.
1.Introduction:
Earlier we had reviewed various aspects of Type 1 Diabetes(T1D)(,its etiopathogenesis,various immunotherapies used and how we could try to obviate the need of insulin ,role of empagliflozin addition ,role of ECV’s in treating complications associated with T1D ,role of gut microbiota and early life feeding,genes responsible (unpublished ),epigenetics in DKD [1-11].Here we decided to conduct a systemic review on role of β cells in etiopathogenesis along with treatment directed towards them.
1.1.Autoimmune Type 1 Diabetes:
Autoimmune Type 1 Diabetes(T1D)(also known as Type 1a Diabetes) occurs secondary to insulin deficit resulting from autoimmune modulated damage of pancreatic β cells[12] Usually it is discriminated from the <common Type 1b Diabetes,or idiopathic /non Autoimmune Diabetes,where insulin deficit and β loss with no β cells Autoimmunity[13].There has been an escalation all over the world in the last few decades[14], as well as although considered a paediatric disease ,recently escalating no of young adults have got diagnosed[1,3].An experience in children as well as adolescents /youth with T1D usually there are problems with insulin dosing for sustainance ideal glycaemic regulation as their age advances.Thus long duration Diabetes- associated complicationslike Diabetic Nephropathy (DN), Neuropathy,retinopathy might be seen in their lifetimes[12].Moreover ,inspite of significantly better insulins ,that have escalated lifespan of people living with T1D,escalating financial barriers are there limiting affordability in a lot of countries [15],with a total greater risk of cardiovascular disease (CVD) ,the major cause of mortalityin T1D people[16] .At present no therapy exists to avoid /cure T1D ,with /day delivery of insulin –only safe ,efficacious managing method .Hence ,besides the clinical care continuing ,an immediate requirement exists for a more extensive T1D pathogenesis as well as generate avoidable treatment as well as curable ones.
Methods:
Thus Here we conducted a systematic review utilizing search engine pubmed,google scholar ;web of science ;embase; Cochrane review library utilizing the MeSH terms like; Type 1 Diabetes(T1D ;beta cell in etiopathogenesis of T1D;Immunotherapies ;role of Unfolded proteins response(UPR);role of senescent β cells ; Role of Type 1 Interferon ;DNA methylation;PDL1 ;Little insulin generation by αcells besides glucagon ;other endocriner cells of pancreas ;Role of autophagy;other mechanisms like apoptosis ;necrosis in β cell demise ; endoplasmic reticulum (ER)stress ;Terminal UPR;Advanced UPR;EM alterations in mitochondria of islet β cell;Endotype;heterogeneity in T1D; of latent autoimmune Diabetes in Adults(LADA) ;Immunotherapies ; β cell therapies ;combination of 2 therapies;DDR;Senolytics;Bcl2;Bcl –XL; circulating cell free DNA (cfDNA) ; Histone mimic suppression of inflammation;BET Inhibitors(Molibresib); Epigenetics modulation of macrophages and β cells; Tauroursodeoxycholic acid(TUDCA), Verapamil(TXNIP inhibitor) Imatinib (IRE1α- ABL inhibitor from 1950 to 2021 till date.
Results:
We found a total of 300 articles out of which we selected 135 articles for this review.No meta-analysis was done.
2.T1D pathogenesis - T1D Stages:
In T1D clinical heterogeneity is believed to occur secondary to various environmental exposures at the time of generation as well as genetic factors ,each of which carry a major part in bringing about β cell autoimmunity[14,19].Marked refining of models utilized for the natural history of T1D has been done in last decades along with consensus view has been generated[18-20].3 separate clinical Stages of disease propagation have been observed,though the time as well as initiation of every stage differs.At the time of the earliest Stage, patients are asymptomatic, as well as due to genetic proneness along with environmental triggers β cell autoimmunity against β cell antigen ,usually insulin[INS], glutamic acid decarboxylase( GAD65) ,Islet antigen2(IA2) as well as ([21]ZnT8).This early asymptomatic stage can antecede a T1D for yrs , as well as escalating amount of autoantibodies associates well with escalated chances of T1D initiation [22]. In case of newly generated risk scores that include genetic ,epidemiological a;long woith immunological factors ,that can markedly anticipate the chance of T1D initiation in children among 2-8yr ages [23]ii) Stage 2 possesses properties of reducing β cell function as well as /or mass as seen by aberrant glucose tolerance test(GTT) as well as in certain instances mild hyperglycemia[12]. Nevertheless, overt hyperglycemia as well as the typical DM symptomatology of polydipsia, polyuria, as well as polyphagia are missing.Recent proof points that β cell impairment instead of totally β cell mass getting depleted during this duration ,might be the key factor for disease propagation[24].Ultimately in the Stage 3, propagation towards becoming totally symptomatic ,in which case which functional β cell mass is not enough to take care of the body metabolic requirements resulting in constant hyperglycemia as well as the typical symptoms of diabetes mellitus (DM) with or without diabetic ketoacidosis.
Intriguingly,a honeymoon duration has been detailed in about 50% of new onset pediatric patients where the symptoms appear to become better as well as clinical remission of DM on the 1st delivery of insulin that was followed by reduction in insulin dosage[25]. Nevertheless, this phase is short lasting as anticipated ,mostly remaining for a few mths as well as patients needing again insulin.This event is not well understood, but might point to avenues for correct timing of treatment to get β cell function retrieved in the long time following diagnosis[26]. Inspite of initially thought that all β cells get damaged in T1D, nevertheless, recent work points that even in well proven T1D(greater than 3yrs following diagnosis ),pro insulin liberation continues for yrs in practically all patients [27] as well as a big part of β cells persist in a lot of patients [28-30].These findings are promising for actions for recovering β cells way following diagnosis .
2.2A Disease implicating immune system as well as β cells-T1D:
T1D has been treated in the form of a Disease implicating immune system[31],in which β cells act as the passive targets that get damaged by a complicated autoimmune event that is modulated by self-reactive cytotoxic CD4+ as well as CD8+ T Cells that gets support via innate immunity.In view of this highlighting,clinical interventions to avoid as well as treat T1D concentrated on immune targeting treatment, certain of which demonstrated advantageous effects[32,33].Like a recent clinical trial utilizing nondepleting antiCD3 antibody(teplizumab),that targets T cells ,in T1D patients relatives who themselves had a great chance of generation of Disease(≥2 autoantibodies as well as initial signs of aberrant glycemia )resulted in a 3yr median postponement in the propagation towards T1D initiation in contrast to placebo[33]. Nevertheless, the precise mode of action of teplizumab are still not known , as well as this antibody thought to be therapeutic further had minimal action in certain patients(like nonresponders)[33].Akin to that a recent trial utilizing golimumab, that is a monoclonal as tumor necrosis factor alpha(TNFα) antibody,resulted in escalation of residual β cell function as well as decreased utilization of insulin in new onset pediatric as well as young adult patients with T1D in contrast to placebo[34].This study further documented an escalated amount of hypoglycemic processes ,besides escalated times of infections in golimumab patients[34] Hence ,whereas certain immunotherapies can postpone propagation of disease at the time of stage2 or even following stage3 initiation,there are certain patients who don’t respond as well as sometimes unanticipated results get encountered following systemic immunomodulation. Lots of immunomotherapy clinical trials for new onset T1D or avoidance of T1D/postponement are ongoing that are immune modulating antibodies, cytokine,vaccines as well as regulatory T cell treatment[35,rev byus ref 4,5].
Generating from the typical posit of T1D as an autoimmune disease, escalating proof points to the thought that β cells impairment is equally key like the autoimmune event, along with T1D being a disease of the β cells or islets [35,36]. Genome –wide association study (GWAS) point that main polymorphisms other than human leukocyte antigen(HLA) complex which have a correlation with T1D are located in genes that we know are expressed in β cells ,that includes INS gene by itself[37].In the last few yrs watching these T1D patients clinically point to the belief that of continuing β cells impairment before the diagnosis, as well as β cells mass as well as function that continues to be present despite the T1D getting established,yrs following diagnosis [16,17,38].Hence a newer stress on β cells drug treatments might become promising method to decrease β cells demise ,get the β cells function back along with avoiding T1D initiation at the time of stage 2 or early into stage 3 of the disease[16].Here some of modes which bring about various types of β cells impairment at the time of stage 2 or stage 3 of T1D initiation as corroborated by mouse as well as human studies,that includes β cells apoptosis, senescence as well as other impaired states with emphasis on clinical translation actions as well as avenues for targeting these particular pathways. Further the probability of combination of β cells drug treatments with immunotherapy for T1D avoidance with the knowledge of continuous reexploration of T1D causation that would be necessary for optimizing the efficacy of every kind of treatments.
2.3β cells impairment in T1D:
2.3A.β cell Endoplasmic Reticulum Stress, Apoptosis,Unfolded Proteins Response:
Probably the best evaluated state of β cells impairment at the time of etiopathogenesis of T1D is endoplasmic reticulum (ER)stress resulting in apoptosis[39,review in 40](figure1A) .Apoptosis by definition is a sort of programmed cell death,that gets triggered by different modes like internally along with irrecoverable cell injury (known as intrinsic pathway),or externally due to surface receptor crosstalk with immune cells(extrinsic pathway) or due to perforin –granzyme pathway[41],[rev in detail ref 42].
Since β cell face great need for insulin development, processing, folding along with liberation, metabolic as well as immune –modulated stress are thought to directly involve the capacity to maintain these events[30]. secondary to this a main etiology of Apoptosis in β cells is ER)stress modulated activation of the loss of Unfolded proteins response(UPR)[43].Hence reduction in Ins1 gene dose enhances β cells ER function just for a little time as well as removes basal UPR stress in mice [44].This UPR represents a 3-branched system which can aid cells to sustain homeostasis(adaptive UPR) or make them to undergo Apoptosis(terminal UPR)[45]. Adaptive UPR signalling aids β cells to meet with the stress of Unfolded /misfolded proteins in the ER as well as recoup ,while a terminal UPR takes place if there is too high or continuous stress,stimulating apoptosis[46](figure 1A).
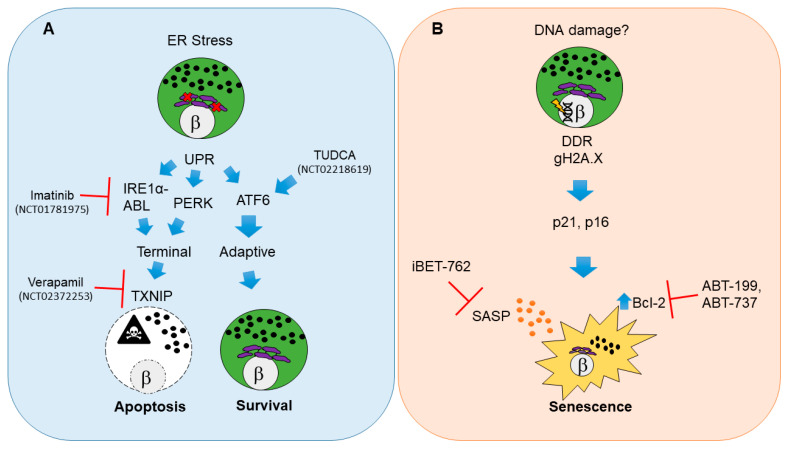
Legend for Figure 1:
Courtesy ref no-40-Molecular pathways and therapeutic targets for beta cell unfolded protein response (UPR)-mediated apoptosis and senescence in type 1 diabetes (T1D). (A) Beta cell apoptosis in T1D results from persistent endoplasmic reticulum (ER) stress that leads to activation of UPR master regulators IRE1α, PERK and ATF6. IRE1α mediates its functions through its RNAse and kinase activities that are potentiated by the Abelson tyrosine-protein kinase (ABLs). The balance of each UPR regulator dictates the outcome on beta cell fate. Unrelieved ER stress signals through IRE1α and PERK and shifts the pathway towards a terminal UPR and apoptosis mediated by thioredoxin interacting protein (TXNIP), whereas ATF6 is the major mediator of adaptive UPR leading to beta cell survival. Clinical trials in new onset adult T1D patients have used Verapamil, Imatinib or tauroursodeoxycholic acid (TUDCA) to attenuate terminal UPR and apoptosis and/or enhance adaptive UPR to delay the decline in residual beta cell function. (B) Beta cell senescence in T1D may be initiated by unresolved DNA damage (although the precise triggers of DNA damage remain unknown). A persistent DNA damage response (DDR) in beta cells is indicated by gH2A.X which is mediated by ATM. DNA damaged beta cells show activation of cyclin-dependent kinase inhibitors p21 and p16, which enforce a senescent growth arrest. Senescent beta cells upregulate the antiapoptotic protein Bcl-2 and develop a senescence-associated secretory phenotype (SASP). Small molecule inhibitors including senolytic compounds targeting Bcl-2 (ABT-199, ABT-737) or suppressing SASP at the level of gene expression (iBET-762) mitigate the deleterious effects of accumulated senescent beta cells in NOD mice and prevent T1D. These drugs have not been tested in clinical trials for T1D. The white circles and the β symbol indicate the nucleus, while the purple structure is the ER and black dots indicate insulin granules.
Despite concentration of recent work on terminal UPR signalling as the main mode that
stimulates β cells apoptosis, proof from the largely evaluated non obese diabetic(NOD) mouse model of human T1D[47].points that β cells undergo apoptosis as well through combination of extrinsic pathway as well as perforin –granzyme pathway that gets directed via cytotoxic T Cells[48,49].Akin to that a lot of studies on human donor pancreatic tissue have validated the thought that β cells get damaged in a heterogenous manner over the pancreas by CD8+ T Cells- modulated cytotoxicity[28,50].
A different type of β cells demise ,pointed to be implicated is necrosis[39],that is a lower type of cells demise occurring secondary go exaggerated injury ,where cells get broken down with intracellular components getting liberated in the extra cellular surroundings ,that stimulates immune activation along with inflammatory responses [51].This is in in contrast to what is believed to take place at the time of apoptosis,since apoptotic cells classically possess v short life as well as get deleted by phagocytosis, resulting in tissue remodeling[39,51].Although necrotic β cells demise appears a lucrative reasoning for the liberation of auto antigen as has been posited [52],the proof for necrotic β cells demise as a mode in T1D remains not conclusive.
One main query in this field involves which kind of β cells demise is the predominant one in T1D, as well as an if β cells demise is usually persistent, relapse-remitting or totally as per the cause as well as based on the situation[53]. Intriguingly,in a recent study utilizing DNA methylation in the form of a biomarkers for circulating cell free DNA (cfDNA) that initiates in β cells observed no proof to validate β cells demise that is ongoing(with death measured as β cells obtained cfDNA in serum)in seroconverted subjects or the ones with recent onset or fully developed T1D,while the same bioassay had great sensitivity to pick up β cells demise seems a promising way following islet transplantation [54]. Hence different kinds of β cells demise during the generation of T1D,whichever they might be ,either vary from the ones during islet transplantation or are just not occurring persistently .With the broadening of our insight into cells demise mode [36] it would be significant to find the extra pathways of β cells demise in T1D.
2.3B. Unfolded proteins response modulated β cells Apoptosis-Pathways:
In case of β cells adaptive as well as , terminal UPR get kept in a balance that is downstream of ER stress. ER stress stimulates the tripartite UPR signalling pathways that implicates the master controllers inositol requiring enzyme -1 alpha(IRE1α),PKR-like ER Kinase(PERK) along with activating transcription factor6(ATF6),every one of that controls the apoptotic vis a vis survival fate outcome[43].Noticeably , mRNA as well as proteins markers of ER stress along with UPR stimulation in β cells are obvious in the initial stage prediabetic NOD mice along with human T1D donor pancreas sections[55].On continuation of ER stress or beyond reproach a transfer from adaptive as well as , terminal UPR through IRE1α or PERK-based stimulation of the redox protein thioredoxin –interacting protein(TXNIP)in β cells [46,56](figure1A).For triggering the intrinsic apoptotic pathway in β cells, TXNIP stimulation is necessary [46,56].As per this , terminal UPR as well as apoptosis in β cells can be avoided utilizing small molecule inhibitors that target the RNAse action of IRE1α or its binding colleague,Abelson tyrosine protein kinase (ABL )[57].Current genetic proof points that IRE1α further regulates β cell identity, as well as β cells particular knockout of this UPR-modulator confers protection against T1D in case of NOD mice[58].Akin to that Txnip knockout avoids the apoptosis getting induced in rodent β cell line as well as islets ex vivo under situations of continuing ER stress,like escalated glucose[59].
3.1. UPR Treatment in T1D-Clinical Trials:
Clinical Trials that were evaluating β cells-aimed treatments for T1D(like where β cells represent the primary target of the experimental substance,excluding transplantation)are occasional.Since November 2020 on the clinicaltrials.gov website there were more than 2100 interventional trials(inclusive of all trial statuses) that are noted,but only roughly 100 of them implicate β cells as targets of drugs,maximum of whom are repurposing agents ,at present being utilized for T2D. Interventional Clinical Trials using small Molecule agents(besides standard insulin regimens) to ameliorate β cells apoptosis in T1D-in adults(≥18yrs old) have demonstrated beneficial initial outcomes in small cohorts .A phase II placebo controlled Trials utilizing daily Verapamil(TXNIP inhibitor) with recent onset Type 1 diabetes in adult subjects >12mths(NCT 02372253) illustrated escalated conservation of β cells function,decreased hypoglycaemic processes as well as reduced insulin needs[60](figure1A). Nevertheless, the size of the study was quite small(n=11 patients for every treatment group) as well as further documented a high rate of GIT adverse actions as well as nausea ,thus it was not clear if the drug might be tolerated Specifically by paediatric population.Akin to that a recent Clinical Trial that utilized Imatinib (IRE1α- ABL inhibitor)in recent onset Type 1 diabetes patients(NCT 01781975) illustrated partial conservation of β cells function in contrast to placebo (unpublished study) ](figure1A). Nevertheless,a wide range of side actions got documented as well as happened more often in the Imatinib-dosage group ,that were clubbed widely as gastrointestinal tract (GIT) ,skin,respiratory ,cardiac,endocrine ,infections that points to a broad category of targets.Actually it was recently documented that besides terminal UPR signalling, Imatinib further directly influences insulin liberation from β cells[61] along with facilitates Reactive oxygen species(ROS)scavenging via B cells in NOD mice,an action that is necessary for reversing of Diabetes[62].Thus it appears one has to gain a lot of further knowledge with regard to this drug.
Terminal UPR as well as apoptosis might further be avoided by escalation of the capacity of β cells to deal with Unfolded proteins. Tauroursodeoxycholic acid(TUDCA),that is a bile acid obtained component works as an ER stress inhibitor along with protein chaperone[63] as well as avoids Diabetes in the NOD mouse model in an ATF6 based way[64] ](figure1A).Noticeably TUDCA –associated acids have been utilized in a safe manner in infants along with children for a little time now in the form of therapy of different hepato-biliary diseases’[65],pointing that they would be safe for the paediatric subjects.A phase II placebo controlled Clinical Trial for TUDCA in recent onset Type 1 diabetes in adult subjects(NCT 02218619)got finished recently ,though outcomes are still to get published.This studies observations would be significant in yielding more Clinical proof for the capacity of UPR inhibitor therapies for escalation of β cell survival as well as function in T1D.Whereas these studies are attractive ,a crucial property of these drug treatments is their need for continuous delivery for hampering their targets as well as be efficacious (daily dosage regimes got utilized in these trials).This type of regimen usually makes the duration along with robustness the maximum.Actually ,the uptake of such UPR hampering drugs in cell kinds other than the ones that are ER stressed pancreatic β cells would be harmful if Terminal UPR as well as apoptosis are needed for tissue regeneration along with cell turnover.However ,the proof from these Clinical Trials point that a definite window for enhancing ,or minimal postpone ,the reduction of the remaining β cells function other than insulin therapy by itself.The query of if β cells function can get enhanced in Type 1 diabetes by repurposing T2D drugs remains still for discussion. Nevertheless, the proof from these Clinical Trials studying glucagon like peptide 1(GLP1), as well as GLP1 receptor signalling point that this might not be efficacious (NCT 01155284, NCTo2284009)[66].With the further studies start to get insight at which time β cells have maximum proneness to ER stress stimulated functional reduction as well as Terminal UPR at the different stages of T1D generation,it might be feasible to utilize these treatments off and on as well as when required maximum ,that prevents the adverse actions that occur following daily delivery.
3.2. Injury – stimulated β cells Senescence:
Even non-lethal types of β cells impairment further aid in T1D generation.A kind of subpopulation of β cells in the late stage of prediabetic NOD mouse ,in seroconverted asymptomatic donors along with recent onset as well as fully generated human T1D donors activate a DNA –damage stimulated senescent fate [67](figure 1 B). Senescence by definition is a kind of programmed growth halt ,usually stimulated by different kinds of unrepairable cellular Injury, aging or oncogenic stimulation [68].Whereas Senescence is typically thought of as a single phenotype /state ,a lot of escalating literature validates the belief the various kinds of Senescence based on cell kind, stage of generation , as well as provoking stimuli[69-71],actions of physiology of the tissue[72,73].Conversely advantageous kinds of Senescence are utilized for a lot of necessary events ,like embryonic growth and patterning[69,74], tissue regeneration[71],wound healing[75] along with tumor suppression[76].Hence, Senescence has been pointed as antagonistic pleiotropy at the time of evolution(i.e where more than 1 trait controlled by a gene where 1 is beneficial in early phase of life while at the late stage is harmful)[77].The absence of a unique marker for Senescence in vivo has seen to it that the correct phenotypic definition of these cells in different tissues very difficult.Hence a lot of independent markers are essential to validate these claims regards to Senescence[78].
The provocateurs of the early DNA –damage as well as Senescence stimulation in β cells at the time of T1D have to be found as yet. However ,the findings that β cells Senescence as well as apoptosis both take place at the time of pathogenesis of T1D in humans as well as mice favours the fact that both represent damage-stimulated fates[79].The queries that need to be addressed are what influences certain β cells to seek terminal UPR ,whereas rest stimulate a damage-associated Senescence program? At the transcriptional as well as functional levels β cells are believed to be heterogenous [80-82], as well as heterogeneity takes place at a lot of level s in T1D[14].Tackling the basic query regarding heterogeneity in β cell fates would be of a lot of significance for getting insight in the pathogenesis of T1D. as well as
3.2B. Injury – stimulated β cells Senescence- Molecular Pathways:
Senescence- associated secretory phenotype (SASP)
With regards to T1D, damage stimulated β cells Senescence display hallmarks of constant DNA –damage response(DDR),that implicates Ser 139 phosphorylated histoneH2A.X (alias gamma H2A.X)[67],that is classically stimulated via the master kinase ataxia telangiectasia mutated(ATM) along with marks-double stranded breaks[83].The halt of growth secondary to senescence gets mediated in these cells by the up regulation of the typical cyclin –based kinase inhibitors , cyclin –based kinase inhibitor1a(Cdkn1a,alias p21) as well as Cdkn2a(that encodes p19Arf as well as p16Ink4a )[67]. Stimulation of ATM mostly signals to stimulate Cdkn1 aexpression through the the p53 tumor suppressor as well as knock out of Atm in β cells ameliorates the DDR which gets stimulated by the DNA –damaging drug streptozotocin [84] that validates the preservation of this pathway in β cells.Noticeably , the kind of β cells Senescence in T1D is separate from what gets seen at the time of age associated β cells Senescence[85], The Senescence as well as T2D[86]. β cells that are aged upregulate p16Ink4a but not p21 as well as don’t display proof of continuing DNA –damage[67,85].The constant DDR of these β cells that are Senescent in T1D further discriminates them from the β cells that are Senescent in T2D visualized in, that simulates an exaggerated aging phenotype[30,86].Moreover it is noticeable that Senescence is not just limited to β cells in T2D , along with the associated metabolic syndrome ,but takes place in a lot of cells that includes preadipocytes as well as hepatocytes’[87].Akin to that Senescence signature in NOD mice was further visualized in human β cells in a small cohort of seroconverted donors(single or double auto antibodies positive)recent onset along with fully Developed T1D donors (that spans <1yr to6yrs disease existence[67].In T1D Senescence in human β cells is seemingly associated with DNA –damage,as validated by the finding that Senescence markers akin to this can get stimulated in normal human islets in culture with the DNA –damaging drug bleomycin [67].Noticeably ,a previous report further illustrated the proof for stimulation of the DDR in β cells of new onset T1D donors (wks to a few mths following diagnosis ),pointed in that study by foci of the factor for repair namely p53 binding proteins 1(53 BP1[85).
Injury – stimulated β cells that are Senescent generate 2 extra phenotypes, Specifically applicable to their harmful actions on the islet microenvironment as well as T1D propagation. Firstly, they particularly upregulate the antiapoptotic protein B Cell lymphoma(Bcl-2)[67](figure1B).The family members of Bcl-2 are either pro or antiapoptotic, that ensures a finely tuned regulation mode over the intrinsic apoptosis[88]. Upregulation of the antiapoptotic protein B Cell lymphoma family members that include extra-large (Bcl-xL), B Cell lymphoma w(Bcl-w) as well as /or Bcl-2 appears to be a main hallmark of maximum kinds of Senescence as well as contributes to a prosurvival phenotype in Senescence as well as cancer [88].Hence β cells that are Senescent can probably dodge the external clues from the environment, that includes lymphocytes that are infiltrating as well as inflammatory macrophages that are resident which in other circumstances would instigate apoptosis. This property in Specific sets, Injury – stimulated β cells Senescence, besides in the form of totally separate fates in contrast to UPR - stimulated apoptosis, since Senescenct β cells have a long life . Second Senescenct β cells can stimulate a pro inflammatory secretome that is classical of other kinds of Senescenct cells which was initially known as Senescence- associated secretory phenotype (SASP) [67,89]. SASP is a relevance –based as well as dynamic program involving secreted cytokines, chemokines, growth factors, shed receptors, as well as matrix proteases which are markedly immunogenic as well as modulate paracrine signalling with the adjacent cells [68,70,90].The basic aim of SASP in vivo appears to be immune surveillance along with removal of Senescenct cell from the tissue resulting in resolving of inflammatory responses[78,90]. Nevertheless,in relevance to TID ,SASP appears to not get resolved since Senescenct β cells keep on collecting as the disease propagates[67]. Senescenct β cells further possess escalated lysosomal β galactosidase activity [67],a phenotype that is shared by β cells aging as well as β cells Senescence in T2D[85,86], known as Senescence- associated β-gal activity[91].
Still one has to find out regarding how transition from Senescence to SASP in β cells occurs,since just a subset of Senescenct β cells generate SASP markers, as well as a lot of difference in the rate of SASP β cells in NOD mice as well as human donors with T1D[67].Lastly it is significant to appreciate whereas these collected Senescenct β cells display changes in certain critical β cells identity genes a(like reduced Ucn3)[67],they are separate from β cells which get totally de differentiated (like illustrating endocrine precursor marker Ngn3) or trans differentiated(like displaying a bi hormonal or poly hormonal phenotype).This type of conclusion gets validated by the findings that they sustain great amount of Ins1 as well as Ins2 expression dependent on single-cell RNA –seq as well as have what looks like normal amounts of insulin as seen by IHC[67].If the Senescenct β cells subpopulation in NOD mice has an overlap with that subset which fights the autoimmune fight as well as continues once fully developed diabetes in this model[92],has to be found ,despite the putative antiapoptotic phenotype of the former agrees with this thought.
4.Senescence Targeting Treatments in T1D-Chances of Clinical Translation:
With the use of pharmacological agents , Senescenct β cells collection can get ameliorated ,resulting in a pause in the autoimmune event as well as avoidance of T1D in NOD mice.Inhibitors of Bcl-2 which act as senolytics(drugs clearing Senescenct cells)agents selectively stimulate in the apoptosis in Senescenct β cells(figure1B) without any change that can get picked up in the main lymphoid or myeloid cell kinds in T1D[67].Hence treatment of islets that have been isolated from NOD mice or delivery of senolytics agents ABT-199 or ABT 737 to,prediabetic mice reduces the Senescencce as well as SASP markers ex vivo as well as in vivo[67].Hence therapy with ABT-199(alias Venetoclax) got recently approval from FDI in the form of 1st class Bcl-2 inhibitor for combination therapies in chronic lymphoid leukemia in which overexpression of Bcl-2 takes place.Akin to that ,suppression of SASP pharmacologically in β cells attained transcriptional inhibition of the bromodomain extraterminal domain(BET) protein family[93].At present small Molecule BET inhibitor iBET[762,at present in phase I/II trials for different cancers[94],avoids diabetes as well as represses SASP in β cells of NOD mice in vivo as well as human islets ex vivo[93].A BET inhibitor from the prior generation iBET-151 was also demonstrated to avoidT1D in NOD mice, as well as pointed actions on both β cells as well as macrophages[95].In toto these observations point that besides BET inhibitors suppress SASP pharmacologically in β cells,they further reduce the BET protein-modulated inflammatory pathways in myeloid cells[96].However,proof from studies in NOD mice,human pancreas donor specimens as well as islet culture models validate the clinical utility in of β cells Senescence therapies for avoidance of T1D.It still is not clear if therapies targeted at Senescence would be advantageous following T1D initiation or might be utilized at the time of partial T1D remission honeymoon phase.
For being successful to get therapies that targeted Senescence in relevance to clinical utility,some problems have to got to be overtaken.1)The present generation of Senescence targeting treatments as well as senolytics are the ones that get repurposed from the oncology branch , as well as whereas maximum possess adverse effects that can be agreeable in adults they have not been evaluated in children ,hence might cause a lot of risk.Open label small cohort phase1 Clinical Trials to delete therapeutically Senescenct cells in adult subjects with Diabetic Kidney Disease[97] or idiopathic pulmonary fibrosis[98] have utilized a cocktail of senolytics agents Dastanib plus Quercetin(D+Q) that are delivered off and on, as well as have illustrated good safety along with some effectiveness . Nevertheless, it is not known if D+Q have the capacity of influencing the, Senescenct β cells collection that occurs in T1D.Secondly ,since these agents possess off target actions, it would be essential to generate targeted administration methods to maximize uptake by Senescenct β cells . Whatever strategies that are coming up regarding therapeutic targeting of Senescenct cells in other tissues [99] might aid in generating a similar system for β cells. 3)Lastly theabsence of Clinical correlates interferes with the capacity of anticipation which seroconverted patients possess the maximum burden of Senescenct β cells as well as thus would get maximum efficacy from these therapies .Actually it appears that a broad difference in the degree of Senescenct β cells in islets of recent onset Type 1 diabetes as well as seroconverted donors[67],that highlights the belief of heterogeneity of β cells fates .Procuring a Biomarker for Senescenct β cells would lay open the stage of questioning patient cohorts to generate association among Senescence along with other clinical features to isolate patients which might prove to be great subjects for β cells Senescence treatment[67].
5.1. Restof States of β cells impairment:Definite proinsulin processing as well as Bihormonal Beta/IsletCells:
A lot of proof for other non-damaged impaired states in Beta Cells has got documented recently .These represent aberrations in proinsulin processing which has been proved in T1D[27,100,101], as well as trans differentiation /changed identity in recent onset as well as generated Type 1 diabetes[102,103]. Proinsulin represents the precursor Molecule subsequent to deletion of the N-terminal signal peptide from pre proinsulin in the ER [104]. Prohormone convertase (PC )1 as well as 3 ,PC2 along with carboxypeptidase E(CPE)that represent neuroendocrine peptidases catalyze stepwise proteolytic cleavage processes which finally develops mature insulin as well as C peptide for exocytosis[105].Noticable studies done by independent workers have illustrated a proinsulin processing impairment in generated Type 1 diabetes,as pointed by i) escalated proinsulin: insulin ratio in islets , as well as ii)constant proinsulin liberation observed in serum of longstanding Type 1 diabetes subjects[27, 100,101], PCSK1 mRNA (that encodes PC 1 as well as 3 isoforms was reduced by significant amount in T1D pancreata ,while expression of PCSK2(that encodes PC 2) as well as CPE were not influenced [101],pointing that the aberration in proinsulin processing occurs due to decrease in PC1 as well as 3 activity. One more study validated this observation at the protein level, with decreased PC1 as well as 3 found from T1D donor islets as well as a pattern towards reduction in CPE amounts [100].Further whereas INS mRNA was plenty in developed T1D pancreata, markedly low nascent transcript(alias heterogenous nuclear RNA ) was observed from the INS promoter ,pointing that transcription that was continuing gets impaired in T1D[101].Then the query comes out that what is the mode by which proinsulin processing impairment starts in long time T1D,that remain significant queries for future.That would aid treatment strategies to enhance proinsulin processing as well as probably insulin generation as well as liberation in longstanding Type 1 diabetes subjects.If this stage of impaired proinsulin processing is a characteristic that occurs along with UPR as well as /or senescence in β cells has to be unearthed.
Besides proinsulin processing impairment,a subset of β cells in recent onset along with longstanding T1D have been illustrated to have a bi hormonal state with simultaneous generation of α cell hormone glucagon along with insulin[102,103].This thought that islet cells trans differentiate in T1D was not corroborated initially till Lam etal.[ 29]stained pancreas specimens obtained from a huge cohort of T1D donors including children to older adults having differing disease time period (from new onset to fully developed ) for islet endocrine markers as well as observed no proof of new β cells generation(alias neogenesis) or bi hormonal islet cells[82]. Nevertheless, another study that was published in the same time duration isolated a highly small sub population(2-5%) of islet cells in a small cohort of fully generated T1D,which were double positive for glucagon as well as insulin,but had absence of canonical alpha cell markers that identify α cell Aristaless related homeobox(ARX) as well as DNA methyl transferase 1(DNMT1)[103]. Following that a better histochemical staining strategy was generated to isolate markedly low amount of insulin expressing cells(insulinlow )in islets from recent onset as well as generated Type 1 diabetes donors ,that were pointed to portray the histological correlate towards the clinical continuation of insulin liberation in micro amounts in case of longstanding T1D[102].Earlier work has pointed that a subset of β cells become insulin negative islet cells might be insulinlow ).
Noticably insulinlow islet cells were isolated in T1D donors of every age ,pointing that this phenotype is not associated with disease period , as well as a subset of these cells in recent onset as well as generated T1D were demonstrated to coexpress islet α cells as well as β cells transcription factor homeobox protein NKX6.1 as well as ARX respectively [102].If these are β cells which have trans differented ,or α cells which had attained low amount insulin generation along with β cells identity markers,could not be found out. Nevertheless, other islet endocrine cells hormones were further documented in the insulinlow cells,like somatostatin, ghrelin as well as pancreatic polypeptide , pointing that insulinlow cells are not generating simply by islet α cells into β cells inter conversion[102].Is it that these cells originate in the asymptomatic stages ,playing an etiological part in T1D etiopathogenesis,or are they generating as a later result of the metabolic actions of as well as suboptimal glycemic regulation?Extra studies are essential to work out how the initiation of insulinlow cells in T1D pancreata as well as find out if insulin generation along with β cells identity can get restored to these cells in T1D subjects .
5.2. Extra modes of β cells impairment:
Various other modes might aid in different ways of β cells impairment,that is not well known ,that includes Viral infections,antiviral responxses as well as impairment in autophagy along with mitochondrial function. Whereas definitive proof for a viral etiology for T1D has to get proved formally [17],a lot of studies have correlated viral infections with T1D[107].Actually a lot of GWAS loci remain in genes possessing antiviral activities that modulate the innate immune signalling through the Type 1 interferon pathway [108]. Antibody modulated repression of the Type 1 interferon signalling avoids T1D in NOD mice [109], as well as treatments that target Type 1 interferon signalling are being utilized to fight a lot of systemic autoimmune diseases [110]. Hyperexpression of HLA Class I takes place at the time of pathogenesis of T1D[111] as well as ahas been associated with Type 1 interferon signalling in human Islet as well as Endo C-β H1 β cells models [112].Polymorphisms in genes that encode innate immune as well as antiviral factors keep a good balance among efficacious host response to Viral pathogens on one end as well as the autoimmunity precipating on the other end[113]. Intriguingly, interferon signalling further facilitates expression of programmed cell death-1 ligand 1(PDL1) on β cells in NOD mice as well as humans [114], a critical immunoprotective factor on β cells [115], hence further interventions to facilitate β cells survival might utilize this pathway.
β cells autophagy is one more significant mode essential for making sure survival occurs at times of stress in mice as well as humans [116]. Impairment in autophagy in β cells of T1D pancreas donors in relation to controls was illustrated in a recent study [117].The other organelles of β cells which might be dysfunctional are mitochondria[118].A recent study pointed no main mitochondrial Ultrastructural changes in β cells in a small cohort of T1D donors by electron microscopy(EM)[119],a new Large –scale electron –microscopy(EM) database for human Type 1 diabetes EM imaging data collection from a much bigger sample of non-diabetics ,autoantibodhy positive as well as T1D donors [120] would be of use for getting the answer for structural impairment in mitochondria of β cells at the time of generation of T1D.
6.β cells treatment Combination with Immunotherapies for Type 1 diabetes Avoidance:
6.1Advantages as well as limitations of Combination treatment strategy for T1D Avoidance:
The thought of Combination of β cells treatment present currently with Immunotherapies for Type 1 diabetes therapy has got recently advised[35],since it seems to be a lucrative strategy for efficaciously tackling impairment o9n either side of the pathogenesis[121](fig2).This idea might implicate treatments which target terminal UPR along with Immunotherapies in Combination like CD3 antibodies,with the aim of escalation of β cells survival as well as reducing or reversing β cells autoimmunity at the time of window following seroconversion as well as early initiation of metabolic impairment(stage2).The more inventions continue on subtle immune as well as metabolic alterations which associate with propagation of seroconversion,the window in the natural history of given a chance for intervening which could avoid subsequent deterioration of functional mass of β cells . ] (figure2).
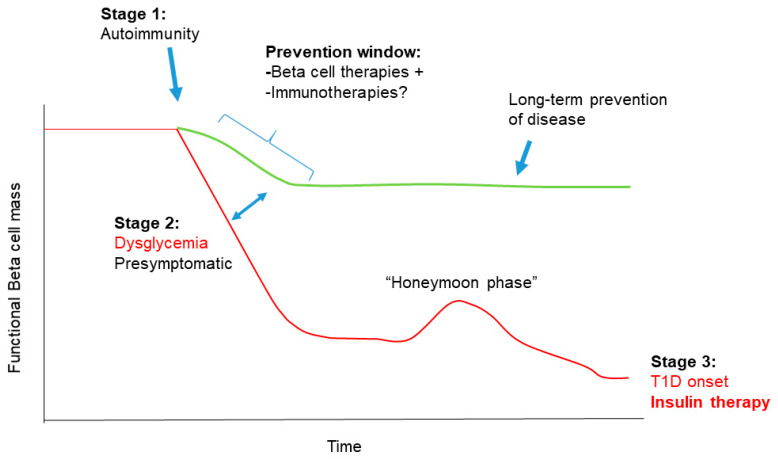
Legend for Figure 2:Courtesy ref no-40-Combining beta cell therapy and immunotherapy for T1D prevention. There is a clear window for preventing T1D onset during stage 2, where seroconversion and dysglycemia are evident but patients are otherwise asymptomatic. The effectiveness of beta cell-targeted therapies, such as drugs inhibiting UPR or targeting senescence could be synergistic with immunotherapy during this stage. Intermittent use and more targeted delivery of these treatments during this preventive window could afford long-term prophylaxis against further loss of beta cell mass and function (green line), altering the typical trajectory of declining beta cell mass and function leading to T1D onset (red line).
Nevertheless, the utilization of Combination treatments would come with its own problems in the clinical scenario.Every one of treatments alone possess a lot of side actions .Thus adding the therapies together would markedly escalate the number of these processes in a particular patients cohort .It is feasible that therapies administered more off as well as on along with greater targeted treatments would ameliorate the adverse actions to certain degree .Furthermore β cells treatments could be delivered in an alternating method with Immunotherapies,since there is no explanation to point that delivering both forms of therapies together would be needed for ideal effectiveness .However ,a further challenge of utilizing Combination treatments in T1D could arise from the nonpanticipation of the actions of 1 treatment on the other cell type (like action of β cells treatments on the immune system.Like ER stress history of UPR Inhibitor imatinib ,that appears to partly postpone deterioration of functional β cells in the new onset T1D(NCT01781975) history of spares β cells to revert T1D in NOD mice[57], along with acting on ROS signalling in B cells ,a property that is essential for its treatment efficacy [62] .Anticipation as well as disengaging the side actions that are not intended regards to β cells treatments on cells in the immune system could thus become a main hindrance for moving further with proof dependent clinical trials utilizing Combination treatments.
6.2B. Combination treatments as well as reanalysis of T1D Etiopathogenesis:
Probably as per context the potential of Combination treatments is the earlier belief made beforehand which looks at T1D being a single uniform disease(even though implicates both immune along with β cells constituents).This belief is gradually getting abandoned ,since escalating knowledge regards to inter patients differences in practically all areas of the disease ranging from epidemiology along with environmental triggers to age of initiation .variations in sex ,degree of autoimmunity robustness , metabolic impairments as well as insulin effectiveness [14,50].Present gaps in what we understand till now regarding T1D etiology area the relative part played by β cells death as well as impairments on one end , as well as immune system impairments on the other end are some aspects in the field where evaluators are trying to critically analyze the earlier presumptions[36].Actually certain researchers in the field getting more serious as well as asking for total reanalysis of T1D causation ,depending on the basis of disease endotypes,implicating mainly immune vis a vis mainly β cells stimulated pathogenesis[121-123].As per this ,already in the field it has been revealed other separate ,albeit poorly grasped types of insulin deficiency –T1D[124],to the lack of β cells auto immunity in idiopathic or nonimmune T1D[2].Certain place among the extreme ,that have characteristics of T1D as well as T2D,is latent autoimmune Diabetes in Adults(LADA)[125],that presents much later in life as well as typical T1D[13]. LADA displays proof of β cells impairments as well as /or deletion in the existence of mild autoimmune generation,that makes it essential for alterations towards classical T1D regimens[126].
Despite still presumptive right now ,collecting both experimental as well as clinical proof in corroborating an endotypic framework in T1D would aid generation of personalized interventions as well as enhancing the efficacy of the clinical trial designs[123].Having this insight ,it would then be feasible to pick up the therapies that are most appropriate depending on the endotype of the patients,that is a big leap moving towards a personalized treatment strategy that has been a long standing dream regarding this disease[123,127]. Nevertheless, despite proper division as per the endotype of the T1D ,early in the natural h/o disease (like at stage 2 ((figure2),in future a more particular strategy of Clinical Trials utilizing a single drug treatment(like β cells or Immunotherapies)that is tailored for the particular instead of trying to influence both β cells as well as immune system utilizing Combination treatments.Anyhow ,since Clinical Trials for treatments that are targeting β cells in T1D are still in budding stage in contrast to the large numbers along with history of Immunotherapies trials [35,128],it is not possible that β cells treatment would get combined with Immunotherapies for avoidance of T1D in the coming future.
7.Conclusions as well as Further Guidance:
This concept that is getting generated of transferring the looking of T1D as just as an autoimmune disease towards a heterogenous disease of both the immune system along with islets,is a significant one that has already aided in newer treatment chances . β cells UPR ,senescence ,proinsulin processing impairments as well as identity alterations are all areas with robust chance for generation of longtime avoidance strategies for the ones at risk of T1D initiation .Actually these stages might be akin to the iceberg tip ,since there exists no explanation to point that no other types of β cells impairments that has to be invented in T1D . Further escalating recalling of impairments in other islet cells like α cells [129] as well as glucagon liberation exocrine atrophy along with pathophysiology[130],that might also yield targets for therapy.
Cytokines play crucial roles in orchestrating complex multicellular interactions between pancreatic β cells and immune cells in the development of type 1 diabetes (T1D) and are thus potential immunotherapeutic targets for this disorder. Lu et al[131] detailed how Cytokines can stimulate controlling functions—like , IL‐10, TGF‐β and IL‐33—are believed to restore immune tolerance and avoid β‐cell damage.As compared to , cytokines like IL‐6, IL‐17, IL‐21 and TNF, that facilitate the differentiation as well as function of diabetogenic immune cells, are thought to lead to T1D onset and progression. However, targeting these impaired cytokine networks does not always result in consistent effects because anti‐inflammatory or proinflammatory functions of cytokines, responsible for β‐cell destruction, are context dependent. Thus, Lu et al ]131] comprehensively summarise the current knowledge on the involvement of well‐known cytokines in both the initiation and destruction phases of T1D, besides explaining the advances in recently discovered roles of cytokines. Additionally, they stressedthe complicated nature as well as involvement of cytokine modulation therapy and detailed the ways in which this strategy has been translated into clinical trials.
Increasing evidence highlights the role of the interleukin (IL)-17 family in pancreatic diseases. IL-17A induces acinar cell injury directly, recruits’ neutrophils, and cooperates with other inflammatory factors to exacerbate pancreatic inflammation. It also triggers islet β-cell apoptosis and nitric oxide-based cytotoxicity, hence exacerbating islet inflammation. IL-17A seems to have different roles in pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer (PC). IL-17A participates in the propagation of acinar-ductal metaplasia (ADM) and PanIN, but not associated with the features of PC stem cells and the overall survival of patients. Acting similar to IL-17A, IL-17B accelerates the invasion and metastasis of PC, and predicts prognosis of PC and the therapeutic effect of gemcitabine.Thus Clarke et al.[132] reviewed the present insight in the pathogenesis of IL-17 in pancreatitis, type 1 diabetes mellitus (T1DM), and PC, as well as potential pharmacotherapy targeting IL-17 and its receptors in pancreatic diseases. The findings summarized in this article are of considerable significance for understanding the essential role of IL-17 in pancreatic diseasesas we had earlier discussed in the role of autoimmune diseases, like endometriosis,RA,SLEetc [133]
The etiology of this disease is complex and difficult to study due to a lack of disease-relevant tissues from pre-diabetic individuals. Yip etal.[134],studied along with conducting gene expression analysis on human pancreas tissues obtained from the Network of Pancreatic Organ Donors with Diabetes (nPOD), and demonstrated that 155 genes were differentially expressed by ≥2-fold in the pancreata of autoantibody-positive (AA+) at-risk individuals in contrast to healthy controls. Only 48 of these genes remained changed by ≥2-fold in the pancreata of fully generatedT1D patients. Pathway analysis of these genes showed a significant correlation with different immune pathways. They could corroborate the differential expression of eight disease-relevant genes by QPCR analysis: A significant upregulation of CADM2, and downregulation of TRPM5, CRH, PDK4, ANGPL4, CLEC4D, RSG16, and FCGR2B was confirmed in the pancreata of AA+ individuals versus controls. Studies have already implicated FCGR2B in the pathogenesis of disease in non-obese diabetic (NOD) mice. Here they demonstrated that CADM2, TRPM5, PDK4, and ANGPL4 were changed akin to the pancreata of pre-diabetic 12-week-old NOD mice compared to NOD.B10 controls, pointing to a possible role for these genes in the pathogenesis of both T1D and NOD disease. The loss of the leukocyte-specific gene, FCGR2B, in the pancreata of AA+ individuals, is particularly interesting, as it may serve as a potential whole blood biomarker of disease progression. To test this, we quantified FCGR2B expression in peripheral blood samples of T1D patients, and AA+ and AA- first-degree relatives of T1D patients enrolled in the TrialNet Pathway to Prevention study. We showed that FCGR2B was significantly reduced in the peripheral blood of AA+ individuals compared to AA- controls. Together, these findings demonstrate that gene expression analysis of pancreatic tissue and peripheral blood samples can be used to identify disease-relevant genes and pathways and potential biomarkers of disease progression in T1D [134]
While many genes associated with the risk of diabetes have been identified to date, the mechanisms by which external triggers contribute to the genetic predisposition remain unclear. Here,Kirak et al.[135] derived embryonic stem (ES) cell lines from diabetes-prone non-obese diabetic (NOD) and healthy C57BL/6 (B6) mice. While overall pluripotency markers were indistinguishable between newly derived NOD and B6 ES cells, we discovered several differentially expressed genes that normally are not expressed in ES cells. Several genes that reside in previously identified insulin-dependent diabetics (Idd) genomic regions were up-regulated in NOD ES cells. Gene set enrichment analysis showed that different groups of genes associated with immune functions are differentially expressed in NOD. Transcriptomic analysis of NOD blastocysts validated several differentially overexpressed Idd genes compared to B6. Genome-wide mapping of active histone modifications using ChIP-Seq supports active expression as the promoters and enhancers of activated genes are also marked by active histone modifications. They further observed that NOD ES cells liberate greater inflammatory cytokines. Their data pointed that the known genetic predisposition of NOD to autoimmune diabetes leads to epigenetic instability of several Idd regions [135].
It is apparent that a lot of queries have to be tackled in this field.Namely the exact association among these β cells impairment states , as well as what initiates β cells to a particular impairment state in any particular islet along with patient? The ones that exist together or are mutually separate? The basic etiologies of every one of them, as well as how it influences the disease pathogenesis in the clinically known stages? Would it be feasible to combine β cells treatment with each other for targeting various types of β cells impairments concurrently? These remain the key queries in this area for tackling in future if our insight of β cells /islet cells impairment in T1D can get safely along with efficaciously translated in clinical scenario.It is certain that our insight of T1D will keep on getting generated as well as get more refined with advances in experimental technological equipment along with strategies ,like high sensitivity immunohistochemistry [102],single cell phenotyping [81],image cytometry as well as high throughput evaluation [28],ultrasensitive hormone assays [27] as well as pancreas slice technology[50]. Nevertheless, propagation will be based on the agreement to challenge the existing dogmas along with long term presumptions regarding T1D[32,122,123].On these bricks the assurance of treatments with objectives of reverting β cells function as well as survival for avoidance along with treatment of T1D will ultimately get achieved.





Open Access By Aditum Open Access Journals id licensed under Creative Commons Attribution 4.0 International License. Based On a Work at aditum.org